
Journal of Gastroenterology Research and Practice
Case Series - Open Access, Volume 6
Rare, aggressive, and elusive: Malignant gastrointestinal neuroectodermal tumor: A case series and review of literature
Aishwarya Karthikeyan*
Jawaharlal Institute of Postgraduate Medical Education and Research, Puducherry, India.
*Corresponding Author : Aishwarya Karthikeyan
Jawaharlal Institute of Postgraduate Medical Education
and Research, Puducherry, India.
Email: aishuhrudaya@gmail.com
Received : May 08, 2026
Accepted : May 25, 2026
Published : Jun 01, 2026
Archived : www.jjgastro.com
Copyright : © Karthikeyan A (2026).
Abstract
Background: Malignant Gastrointestinal Neuroectodermal Tumor (GNET), also called clear cell sarcoma-like tumor of the gastrointestinal tract, is a rare, aggressive mesenchymal neoplasm with distinctive morphology, immunoprofile, and EWSR1 rearrangements. Its diagnosis is challenging due to overlap with other neoplasms.
Case reports: Case 1: A 29-year-old woman with chronic iron deficiency anemia presented with two months of abdominal pain and severe anemia (hemoglobin 2.9 g/dL). CT showed a heterogeneously enhancing distal jejunal mass with aneurysmal dilatation, mesenteric lymphadenopathy, and hepatic lesions. Segmental resection revealed a solid grey-white tumor involving the jejunal wall. Histology showed cords, nests, and trabeculae of monomorphic round cells with vesicular nuclei, permeative borders, and necrosis. IHC demonstrated SOX10 positivity, patchy S100 positivity, and negativity for cytokeratin, CD117, HMB-45, desmin, WT1, and chromogranin. FISH confirmed EWSR1 rearrangement. The postoperative course was complicated by hollow viscus perforation; the patient was subsequently lost to follow-up. Case 2: A 45-year-old man presented with abdominal pain and intestinal obstruction. CT revealed an endophytic anal canal mass. Biopsy showed a subepithelial tumor composed of epithelioid polygonal cells with clear-to-eosinophilic vacuolated cytoplasm and round-to-oval nuclei, without melanin pigment or necrosis. IHC demonstrated strong diffuse S100 and SOX10 positivity, with negativity for cytokeratin, HMB 45, Melan-A, CD68, CD117, CD56, and synaptophysin. FISH confirmed EWSR1 rearrangement. The patient was lost to follow-up after biopsy.
Conclusion: GNET should be considered in the differential diagnosis of gastrointestinal tumors that are S100- and SOX10-positive but negative for melanocytic markers. Accurate diagnosis requires correlation of histologic, immunohistochemical, and molecular features.
Keywords: EWSR1; Gastrointestinal neuroectodermal tumor; Clear cell sarcoma-like tumor; HMB45; SOX10.
Citation: Karthikeyan A. Rare, aggressive, and elusive: Malignant gastrointestinal neuroectodermal tumor: A case series and review of literature. J Gastroenterol Res Pract. 2026; 6(3): 1254.
Introduction
Malignant Gastrointestinal Neuroectodermal Tumor (GNET), otherwise known as Gastrointestinal clear cell sarcoma, is a rare and aggressive primary gastrointestinal malignant mesenchymal tumor [1]. Clear Cell Sarcoma (CCS) typically arises in the tendons and aponeuroses and was initially described in 1965 by Enzinger as Clear Cell Sarcoma of the Tendons and Aponeuroses (CCSTA) and was renamed by him in 1983 to malignant melanoma of soft parts. However, the first ever documented case of CCS of the gastrointestinal tract was by Ekfors et al. in 1993 [2]. There is no defined incidence rate in any cancer registries; however, although not much, a few case series and reports do not cross a hundred.
It is usually seen in adolescents and young adults with equal gender distribution. The most common site is the small bowel, followed by the stomach, colon, and peritoneum. It is defined as a sarcoma involving the GI tract with neuroectodermal differentiation and gene fusion translocations involving EWSR1, usually EWSR1-ATF1 or EWSR1-CREB1 [1].
Histopathologically, these tumors have epithelioid/ and or spindle cell morphology and can be easily misdiagnosed as one of the more common tumors of the GIT, such as GIST, Neuroendocrine Tumor (NET), Malignant Peripheral Nerve Sheath Tumor (MPNST), melanoma, spindle cell sarcomas, and synovial sarcomas. Due to the aggressive nature of GNET and distinct pathogenesis, prognosis, and treatment, it is essential to distinguish these entities. Its extreme rarity and diagnostic complexity pose a clinical conundrum. Herein, we present two cases of GNET with varied presentations.
Case 1
A 29-year-old female, a known case of iron deficiency anemia on therapy, presented with insidious onset abdominal pain persisting for two months. On presentation, she had a hemoglobin of 2.9 g/dl. There were no signs of active bleeding. Contrast-Enhanced Computed Tomography (CECT) showed a heterogeneously enhancing, irregular thickening involving the distal jejunum, with associated aneurysmal dilatation of the affected bowel segment (Figure 1A). The thickened segment measures 11.7 cm in length and has a maximum thickness of 4.5 cm. In addition, there were multiple enlarged, heterogeneously enhancing perilesional and mesenteric lymph nodes, and two hypo-enhancing hypodense lesions were seen in the liver segments 4A and 4B. The patient underwent resection of the bowel segment.
A solid grey-white growth measuring 11×0.5×6 cm arising from the mesentery was noted on macroscopic examination of the resected specimen. It was seen involving the wall of the small intestine, causing ulceration. Histological examination revealed a tumor arranged in cords, nests, trabeculae, and focal sheets (Figures 1B & 1C). These tumor cells are predominantly monomorphic with round monomorphic nuclei, vesicular chromatin, and inconspicuous but occasionally prominent nucleoli. Admixed are areas of necrosis. The tumor has permeative borders and involves the lamina propria and muscularis layer of the jejunum. Immunohistochemistry reveals that these tumor cells are negative for pan-cytokeratin, ruling out carcinoma, and negative for CD117, excluding Gastrointestinal Stromal Tumors (GIST). They are also negative for HMB45 (Figure 1F), eliminating the possibility of melanoma or perivascular epithelioid cell tumor (PEComa), and negative for desmin, ruling out a skeletal muscle origin. Additionally, the cells are negative for WT1, excluding Desmoplastic Small Round Cell Tumor (DSRCT), and chromogranin, with patchy positivity for synaptophysin and CD56, lowering the likelihood of a neuroendocrine tumor (Figures 1G & 1H). The tumor cells exhibit diffuse positivity for vimentin and SOX10 (Figure 1D) and patchy positivity for S100 (Figure 1E). On FISH, the sample showed tumor cells that are positive for EWSR1 rearrangement (80/100 cells).
Post resection, on postoperative day 15, the patient developed complaints of fever and abdominal pain with increased drain output. On CECT, there was evidence of hollow viscus perforation and she was managed accordingly. Furthur, the patient was lost to follow-up.
Case 2
A 45-year-old man presented to the emergency department with abdominal pain and intestinal obstruction. CT imaging revealed an endophytic mass in the anal canal protruding into the lumen. For further investigation, a biopsy was sent to the histopathology department. However, after the biopsy, the patient was lost during follow-up.
Histologically, the tumor was located in the subepithelium. It was predominantly epithelioid in morphology and composed of polygonal cells with clear to eosinophilic vacuolated cytoplasm and round to oval, centrally placed nuclei with inconspicuous nucleoli. Occasional mitotic figures were noted along with thin walled blood vessels throughout the tumor. There were no areas of gland formation, melanin pigment, or necrosis seen in the tumor (Figures 2A & 2B).
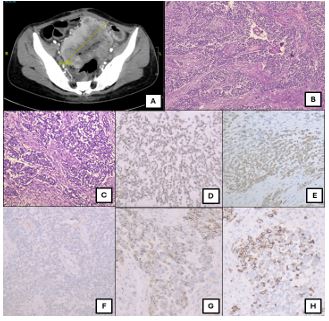
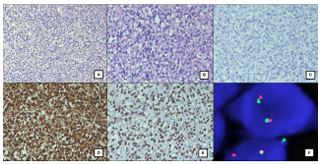
Table 1: Summary of the case reports published in the past five years (2021 - 2025).
| Publication | Year | Country | Site | Age | Sex | Metastasis | Molecular findings | Follow up |
|---|---|---|---|---|---|---|---|---|
| Kumar et al. [13] | 2020 | India | Sigmoid colon | 28 | F | - | FISH: t(12;22) (q13;q12)translocation | Regular follow-up |
| Harshavardhini et al.[14] | 2020 | India | Ileum | 33 | M | - | FISH: EWSR-1rearrangement | Adjuvant chemotherapy, no follow-up |
| Mercedes Bravo-Taxa et al. [15], 2 cases | 2020 | SouthAmerica | 6070 | MF | - | FISH: EWSR-1rearrangement (1) | - | |
| Huang et al. [16] | 2020 | China | 57(34-81) | 2M, 2F | - | EWSR1 mutation | - | |
| Bai et al. [17] | 2020 | China | Rectum | 54 | F | lymphnode, lung,liver, bone | FISH:EWSR1-rearrangement | Thepatient received toripalimab. Dieda few weekslater |
| Okada et al. [18] | 2020 | Japan | Small Intestine | 38 | F | Liver | FISH: EWSR1gene fusion | Liver metastasis after 3 years |
| Sbaraglia et al. [19] | 2020 | Italy | Base of tongue | 62 | F | lymphnode | NGS: EWSR1-CREB1fusions | Doing well at 4 months. |
| Nachiappan M et al. [20] | 2021 | India | Jejenum | 26 | M | peritoneum, omentum, liver, lymph nodes,pleura | NGS: EWSR1-CREB1fusions | Recurrencein just 4 months with worsening comfort care and death. |
| Liao et al. [21] | 2021 | China | Stomach | 49 | F | Liver | - | Recurrence after10 months |
| Zhu et al. [22] | 2021 | China | Ileum | 65 | M | - | FISH, NGS: EWSR1-ATF1fusions | Doing well at 7 months |
| Mishra et al. [23] | 2022 | India | Small Intestine | 32 | M | - | (FISH) demonstrating the presence of EWSR1 (22q12)translocation | The patientis disease-free for 20months of postsurgery. |
| Kandler et al. [8] 20 cases | 2022 | Canada | Small Intestine (9), Liver (5),Soft tissue (4), Appendix (1),Stomach (1) | - | FISH: EWSR1-ATF1 (10)EWSR1-CREB1 (6)EWSR1-FLI1 (2) | - | ||
| Youssef et al. [24] | 2022 | USA | Stomach | 71 | M | Liver, spleen | EWSR1 gene rearrangement | Metastasis: 2months; died in a week |
| Sasaki et al. [25] | 2022 | Japan | Small Intestine | 69 | M | Lymph nodes | (ESWR1) generearrangements | No follow-up |
| Huang et al. [26] | 2022 | China | Distal ileum | 16 | M | LN, liver,lung, bone, left thigh, pleura, adrenal | - | Survival timewas 4 years and 8 months |
| Jiang et al. [27] | 2023 | China | esophagus | 26 | F | Vertebra | NGS: EWSR1-CREB1fusions | On follow-up |
| Shalaby et al. [28] | 2023 | Oman | Jejenum | 34 | F | - | WES: EWSR1::ATF1; DOG1expression | Adjuvant chemotherapy |
| Boșoteanu et al.[29] | 2023 | Romania | Jejunum | 67 | M | - | - | Deteriorated in 6 weeks |
| Nijma et al. [30] | 2024 | Tunisia | Ileum | 20 | F | lymphnode | Not done | Metastasis after11 months |
| Fournier et al. [31] | 2024 | Canada | Duodenum | 73 | F | FISH: EWSR1-CREB1translocation | Regular follow-up | |
| She et al. [32] | 2024 | USA | Small Intestine | 32 | M | Liver (lymphoma& UC) | FISH: EWSR1-ATF1rearrangement | Patient optedfor comfort care. |
| Jia et al. [33] | 2024 | Canada | Terminal ileum | 49 | M | Lymph nodes | EWSR1/ATF1 fusion | Died after15 months |
| Rotaru et al. [34] | 2024 | Romania | Jejenum | 44 | M | - | NGS: Alteration to the MYC, STAG2and EWSR1 genes,EWSR1-CREB1fusion | No adjuvant therapy. On regular follow-up |
On immunohistochemistry, these tumor cells were strongly and diffusely positive only for S100 (Figure 2D) and SOX10 (Figure 2E). The tumor cells were negative for Cytokeratin, HMB-45, Melan A (Figure 2C), CD138, CD68, CD21, CD23, CD117, CD56 and Synaptophysin. A diagnosis of GNET was considered and confirmed on FISH. FISH examination revealed EWSR1(22q12) translocation rearrangement in 61/100 cells (Figure 2F).
Discussion
CCS of the GIT (CCSGT) was first described by Zambrano et al. in a series of six tumours, as an entity distinct from CCSTA despite a few morphological overlaps. The distinct features include the presence of osteoclast-type giant cells and lack of expression of melanocytic markers. This led them to propose a new entity, ‘osteoclast-rich tumour of the GIT with features resembling CCS of soft parts. More research down the lane, revealed evidence of neuroectodermal differentiation, hence the term ‘malignant Gastrointestinal Neuroectodermal Tumour (GNET)’, which is currently a widely accepted terminology.
It occurs in young and middle-aged adults with a slight preponderance of females. The most common presentation encountered is abdominal pain followed by intestinal obstruction. In many instances, it was an incidental finding [3 5]. The most common site of presentation being small intestine, followed by stomach and large intestine [3]. The largest case series was from Stockman et al. who reported 16 cases of GNET followed by Bin et al with 19 cases. It is reported to have distinct morphological feature, clinical treatment and response, particularly to targeted therapy [3,6].
It is typically detected in the gastrointestinal wall epic entered to the muscularis propria and seen extending into the submucosa and/or subserosa [2,7]. Histologically they can be epithelioid or spindle shaped and are composed of monomorphic medium sized to large sized ovoid or epithelioid cells with pale eosinophilic to clear vacuolated cytoplasm. Some can also show pseudopapillary, pseudoalveolar, microcystic, or trabecular pattern. They show frequent mitotic figures. Multinucleated giant cells although said to be a characteristic feature of GNET by some authors, a few have differed in this feature, with only 1 case in series of 19 cases of Chang et al showing this feature [3]. Similarly, our cases showed epithelioid morphology with clear to eosinophilic cells with the appearance of a mesenchymal neoplasm. We did not notice any giant cells, melanin pigment or gland formation.
GNET shares morphological similarities to CCSTA however lacks melanocytic differentiation which is evident on electron microscopy. On electron microscopy, GNET shows features of neural differentiation, including multiple interdigitating processes containing dense-core granules and clear vesicles having synaptic bulbs but lacks evidence of melanocytic differentiation. Although, the exact origin is uncertain, these features point towards the possibility of origin from an autonomic nervous system related primitive cell of neural crest [6]. This is reflected on immunohistochemistry which shows neural or neuroectodermal markers such as S100, SOX10, NSE and CD56 with absence of melanocytic differentiation (HMB45, Melan-A).
The differential diagnosis for GNET includes the most common mesenchymal tumor in all parts of GIT which is Gastrointestinal Stromal Tumor (GIST). Further, in the esophagus and anal canal: GIST, carcinosarcoma and melanoma. The other differentials have been considered in various instances are MPNST, neuroendocrine tumors, spindle cell liposarcoma, undifferentiated sarcoma, monophasic synovial sarcoma, carcinosarcoma and lymphomas.
The differentiation from these tumors becomes crucial due to the very aggressive nature and distinct treatment profile of GNET. The most difficult differential diagnosis lies with CCSTA involving the GIT (“true” clear cell sarcoma). They have similar histological appearance, S100 expression and shared molecular translocation. In order to separate the two entities, the lack of melanocytic differentiation has been reported as a feature of CCSLGT to distinguish it from CCS or melanoma. The expression of S100 protein varies from diffuse to patchy and melanocyte specific markers such as Melan-A and HMB-45 are not expressed. This was the differentiating feature seen in the present case too. Some cases show expression of neuroendocrine markers such as Chromogranin-A, Synaptophysin, CD56 and Neuron Specific Enolase (NSE). The first case that we have described, demonstrated strong and diffuse positivity of S100 and SOX 10 with focal positivity for other markers like CD56 and synaptophysin. CD56 positivity has been demonstrated in a few cases in literature and is positivity attributed to the neuronal differentiation.
On the molecular front, these tumor harbor EWSR1 chromosomal translocations, the most common one being EWSR1-ATF1 and EWSR1-CREB1. It is generally thought that EWSR1 fusions with various partner genes occurs early and serves as a molecular driver in the process of oncogenesis. These translocations are seen in around 86 to 93% of the cases [3,6]. An elaborate genomic analysis of 20 cases by Kandler et al revealed translocations including EWSR1-ATF1, EWSR1-CREB1 and EWSR1-FLI1 fusions with introns 7 and 8 being the common break points noted in EWSR1. In addition, the other genomic alterations noted are copy number loss in CDKN2A/B and short variants ASXL1, BCOR, BCORL1, CREBBP, ECT2L, MAGI2, TP53, SETD2 and RARA [8].
EWSR1 rearrangements are not exclusive to GNET and occur in various other tumors like conventional CCS, Ewing sarcoma, hyalinizing clear cell carcinoma of salivary gland, myoepithelial carcinoma, extraskeletal myxoid chondrosarcoma, myxoid liposarcoma, angiomatoid fibrous histiocytoma and desmoplastic round cell tumors [5,9]. Most of these tumors are malignant which implies that EWSR1 gene rearrangement may be involved in the pathogenesis but is not a specific molecular abnormality. In present cases too we got an EWSR1 rearrangements.
GNET represent a highly malignant neoplasm with poor prognosis and aggressive clinical course. However, there are reports with variable prognosis. In the series described by Stockman et al, a large proportion of patients presented with lymph node and liver metastases (7/16, 43.6%) [6]. In another large series by Chang et al. 5/17 patients presented with metastasis (1 to liver and 4 to lymph nodes) [3]. Li et al. did a thorough clinical outcome analysis from 96 reported cases published as of 2020. The study proved that the Overall Survival (OS) varied from 0.69 to 161 months, and the median OS was 61 months. Most notably, eight patients had long survival times with more than five years [10].
The effectiveness of chemotherapy and targeted agents in treating GNET (gastrointestinal neuroectodermal tumors) is still unknown and unpredictable. For early-stage disease, adjuvant or neoadjuvant chemotherapy has been infrequently employed, with sparse case reports only, and the results infrequently reported because of loss of follow-up [11]. One patient achieved long disease-free survival following platinum based chemotherapy. Different sarcoma-based regimens have been experimented with mixed results [10]. In metastatic GNET, systemic chemotherapy in general has resulted in poor response, but partial responses or stable disease have occurred in some individual cases. Targeted therapy including mTOR inhibitors and multityrosine kinase inhibitors (e.g., crizotinib, pazopanib) have been employed in a few individual cases with mixed, frequently brief effects. Combined TKI therapy led to durable response in one patient, possibly on the basis of c-MET inhibition [12]. Radiation therapy is unproven to be effective but potentially useful to manage symptoms in the metastatic context, especially in bone metastases [10].
Conclusion
Gastrointestinal Neuroectodermal Tumor (GNET) is an uncommon and aggressive mesenchymal malignancy that must be included in the differential diagnosis of tumors of the gastrointestinal tract wall, especially those with spindle or epithelioid morphology and diffuse S100 positivity but without overt melanocytic differentiation. Due to the diagnostic difficulty and shared features with other neoplasms, verification by molecular analysis for EWSR1 gene rearrangements is necessary. Our case series underscores the need to incorporate histopathology, immunohistochemistry, and molecular diagnostics to properly diagnose GNET and direct clinical management.
Informed consent
Written, informed consent was obtained from both patients for publication of the case details and any accompanying images
References
- BlueBooksOnline. 2025. Available from: https://tumourclassification.iarc.who.int/chaptercontent/31/216
- Ekfors TO, Kujari H, Isomäki M. Clear cell sarcoma of tendons and aponeuroses (malignant melanoma of soft parts) in the duodenum: The first visceral case. Histopathology. 1993; 22: 255–260.
- Chang B, Yu L, Guo WW, Sheng WQ, Wang L, Lao I, et al. Malignant gastrointestinal neuroectodermal tumor: Clinicopathologic, immunohistochemical, and molecular analysis of 19 cases. Am J Surg Pathol. 2020; 44: 456–466.
- Morani AC, Ramani NS, Yedururi S, Prasad SR. Malignant gastrointestinal neuroectodermal tumor: A new kid on the block? J Comput Assist Tomogr. 2022; 46: 676–682.
- Thway K, Fisher C. Tumors with EWSR1-CREB1 and EWSR1-ATF1 fusions: The current status. Am J Surg Pathol. 2012; 36: e1–e11.
- Stockman DL, Miettinen M, Suster S, Spagnolo D, Dominguez-Malagon H, Hornick JL, et al. Malignant gastrointestinal neuroectodermal tumor: Clinicopathologic, immunohistochemical, ultrastructural, and molecular analysis of 16 cases with a reappraisal of clear cell sarcoma-like tumors of the gastrointestinal tract. Am J Surg Pathol. 2012; 36: 857–868.
- Sonai MK, Rastogi S, Madhusudhan KS, Gupta SD, Das P. Clear cell sarcoma-like tumor of gastrointestinal tract: Experience of three cases and review of literature. Indian J Pathol Microbiol. 2020; 63: 90–95.
- Kandler T, Cortez E, Clinton L, Hemmerich A, Ahmed O, Wong R, et al. A case series of metastatic malignant gastrointestinal neuroectodermal tumors and comprehensive genomic profiling analysis of 20 cases. Curr Oncol. 2022; 29: 1279–1297.
- Konstantinidis A, Cheesman E, O’Sullivan J, Pavaine J, Avula S, Pizer B, et al. Intracranial angiomatoid fibrous histiocytoma with EWSR1-CREB family fusions: A report of 2 pediatric cases. World Neurosurg. 2019; 126: 113–119.
- Li R, Cao J, Chen L, Cui F, Chen S, Feng Z, et al. Malignant gastrointestinal neuroectodermal tumors: Clinicopathological and prognostic features of 96 patients. Onco Targets Ther. 2020; 13: 9731–9740.
- Singh D, Atieh MK, Russell MA, Kittaneh M. Malignant gastrointestinal neuroectodermal tumor (GNET) with prolonged disease-free survival after platinum-based chemotherapy. Case Rep Oncol Med. 2020; 2020: 8880202.
- Subbiah V, Holmes O, Gowen K, Spritz D, Amini B, Wang WL, et al. Activity of c-Met/ALK inhibitor crizotinib and multi-kinase VEGF inhibitor pazopanib in metastatic gastrointestinal neuroectodermal tumor harboring EWSR1-CREB1 fusion. Oncology. 2016; 91: 348–353.
- Kumar N, Bhoriwal S, Yadav R, Deo SVS. Clear cell sarcoma of sigmoid colon: A rare malignancy with common clinical manifestations. J Gastrointest Cancer. 2020; 51: 1070–1073.
- Harshavardhini S, Saishalini CN, Pavithra V, Shah NM, Sankar S. Malignant gastrointestinal neuroectodermal tumor: A case report. Indian J Pathol Microbiol. 2021; 64: 373–375.
- Bravo-Taxa M, Huanca-Amesquita L. Malignant gastrointestinal neuroectodermal tumor: A report of 2 cases and a review of the literature. Rev Esp Patol. 2022; 55: 267–273.
- Huang HJ, He YH, Fan DG, Chen XY. Malignant gastrointestinal neuroectodermal tumor: Clinicopathological analyses of four cases. Zhonghua Bing Li Xue Za Zhi. 2020; 49: 821–826.
- Bai C, Dong M, Shen W. Rectal clear cell sarcoma: A case report. Transl Cancer Res. 2020; 9(10). Available from: https://tcr.amegroups.org/article/view/44113
- Okada T, Hirano Y, Ishikawa S, Kondo H, Ishii T, Yamaguchi S. A long-term survivor of clear cell sarcoma-like tumor of the gastrointestinal tract with liver metastasis: A case report. Surg Case Rep. 2020; 6: 260.
- Sbaraglia M, Zanatta L, Toffolatti L, Spallanzani A, Bertolini F, Mattioli F, et al. Clear cell sarcoma-like/malignant gastrointestinal neuroectodermal tumor of the tongue: A clinicopathologic and molecular case report. Virchows Arch. 2021; 478: 1203–1207.
- Nachiappan M, Srikantaiah GD, Gadiyaram S. Clinical, pathological, and genetic profile of clear cell sarcoma-like tumour of jejunum: Report of a rare aggressive tumour of small bowel. Clin J Gastroenterol. 2022; 15: 107–111.
- Liao S, Wang X, Li J, Yu X. Clinical presentation and imaging characteristics of clear cell sarcoma-like tumour of the gastrointestinal tract with liver metastasis: A case description. Quant Imaging Med Surg. 2021; 11: 4690–4694.
- Zhu P, Zhang T, Bi K, Wu Y, Chen X, Zhang H, et al. Primary clear cell sarcoma of the ileum: A case report with next-generation sequencing analysis. Int J Surg Pathol. 2021; 29: 677–684.
- Mishra P, Biswas D, Pattnaik SA, Patra S, Muduly DK, Balasubiramaniyan V, et al. Malignant gastrointestinal neuroectodermal tumor: A case-based review of literature. J Cancer Res Ther. 2022; 18: 885–889.
- Youssef B, Mohamed RM, Vahhabaghai P, Asberry D. An incidental malignant gastrointestinal neuroectodermal tumor of the stomach: A rare case report and a literature review. Cureus. 2022; 14: e28042.
- Sasaki M, Tanaka M, Asukai K, Koguchi H, Inoue Y, Moriyama M, et al. Malignant gastrointestinal neuroectodermal tumor presenting with small intestinal obstruction: A case report. DEN Open. 2022; 2: e119.
- Huang WP, Li LM, Gao JB. Postoperative multiple metastasis of clear cell sarcoma-like tumor of the gastrointestinal tract in adolescent: A case report. World J Clin Cases. 2022; 10: 6175–6183.
- Jiang DX, Song Q, Liu J, Hou YY. Primary gastrointestinal clear cell sarcoma/malignant gastrointestinal neuroectodermal tumor of esophagus with thoracic vertebral metastasis: Report of a case. Zhonghua Bing Li Xue Za Zhi. 2023; 52: 730–733.
- Shalaby A, Telugu RB, Deshpande PA, Qureshi A, Al Adawi H, Al Harthi S, et al. Malignant gastrointestinal neuroectodermal tumor of small intestine showing DOG1 expression: A case report and review of literature. Int J Surg Pathol. 2024; 32: 374–379.
- Boșoteanu M, Cristian M, Așchie M, Baz RA, Zielonka AM, Cozaru GC, et al. The malignant gastrointestinal neuroectodermal tumor (GNET): A distinct entity and the challenging differential diagnosis with mesenchymal, lymphoid, and melanic tumors: A case report and brief review of the literature. Diagnostics (Basel). 2023; 13: 1131.
- Njima M, Lahbacha B, Ben Jabra S, Moussa A, Bellalah A, Ben Abdeljelil N, et al. Small intestine gastrointestinal clear cell sarcoma: A case report and review of the literature. J Investig Med High Impact Case Rep. 2024; 12: 23247096231225869.
- Fournier A, Deslauriers V, Giguère CC, Borduas M, Collin Y. Malignant duodenal gastrointestinal neuroectodermal tumor (GNET): Case report and review of the literature. Int J Surg Case Rep. 2024; 123: 110195.
- She T, Ren S, Katz S. Gastrointestinal neuroectodermal tumor/extraskeletal Ewing sarcoma of the ileum with ulcerative colitis. Case Rep Gastroenterol. 2024; 18: 449–453.
- Jia Y, Yan Y, Hebbard P, Garvin G, Lu MV. Malignant gastrointestinal neuroectodermal tumor (GNET) mimicking small bowel lymphoma: A case report. Cureus. 2024; 16: e59105.
- Rotaru V, Chitoran E, Mitroiu MN, Ionescu SO, Neicu A, Cirimbei C, et al. Intestinal clear cell sarcoma: A case presentation of an extremely rare tumor and literature review. Medicina (Kaunas). 2024; 60: 847.
- Ulici V, Hornick JL, Davis JL, Mehrotra S, Meis JM, Halling KC, et al. Extraenteric malignant gastrointestinal neuroectodermal tumor: A clinicopathologic and molecular genetic study of 11 cases. Mod Pathol. 2018; 31: 694–706.