
Journal of Gastroenterology Research and Practice
Research Article - Open Access, Volume 5
Systematic review: Clinicopathologic features of gastrointestinal involvement in different amyloidosis forms
Bipneet Singh1*; Palak Grover1; Jahnavi Ethakota1; Sakshi Bai1; Gurleen Kaur2; Rahul Jain3; Merritt Bern1
1Henry Ford Jackson, Health System Located in Jackson, Michigan, USA.
2Government Medical College, Amritsar, India.
3Sri Manakula Vinayagar Medical College and Hospital, Puduchery, India.
*Corresponding Author : Bipneet Singh
Henry Ford Jackson, Health System Located in Jackson, 205 N East Michigan Ave, JACKSON, MI 49201, Michigan, USA.
Tel: 5174997929;
Email: drbipneetsingh18@gmail.com
Received : Jun 05, 2025
Accepted : Jun 23, 2025
Published : Jun 30, 2025
Archived : www.jjgastro.com
Copyright : © Singh B (2025).
Abstract
Gastrointestinal (GI) involvement in amyloidosis is a recognized but relatively uncommon manifestation that presents variably across different amyloid types. This review synthesizes data from multiple studies to delineate amyloid subtypes affecting the GI tract, associated clinical symptoms, diagnostic findings, and systemic involvement.
We analyzed seven studies conducted between 1993 and 2021 involving biopsy-proven GI amyloidosis. Studies included both localized and systemic cases with Gastrointestinal (GI)- dominant features. Data were extracted on the distribution of amyloid types, clinical manifestations, histological sites of deposition, systemic involvement, and survival outcomes.
Patients had predominantly the AL type of amyloidosis associated with gastrointestinal involvement. Diarrhea was the most predominant symptom. Histology was variable, ranging from polypoid to erosive lesions. People often had systemic, kidney, or heart involvement.
GI amyloidosis is a rare manifestation, often overlooked due to paucity of specific. Diagnosis requires vigilance by the physician, endoscopic study, and biopsy prompted by a pre-existing systemic diagnosis.
Keywords: Amyloidosis; Amyloid light-chain; Reactive amyloidosis; Amyloid transport protein transthyretin; Congo red.
Citation: Singh B, Grover P, Ethakota J, Bai S, Kaur G, et al. Systematic review: Clinicopathologic features of gastrointestinal involvement in different amyloidosis forms. J Gastroenterol Res Pract. 2025; 5(2): 1229.
Introduction
Amyloidosis is a disorder characterized by the abnormal folding of proteins, leading to their formation of amyloid fibrils, which deposit in tissues. This process disrupts normal organ structure and function. A variety of clinical syndromes, such as cardiomyopathy, nephropathy, macroglossia, and neuropathy, are caused by amyloid buildup in the respective organs [1,2].
The most prevalent form of systemic amyloidosis is Amyloid Light-chain (AL) amyloidosis, which is linked to plasma cell dyscrasias and the presence of monoclonal light chains in the serum or urine. It is the predominant type affecting the Gastrointestinal (GI) tract. The overall 4-year survival rate for AL amyloidosis ranges from 40% to 60%, but early mortality remains high, largely due to severe cardiac involvement, with nearly one-third of patients dying within the first year after diagnosis. Reactive Amyloidosis (AA), the most common type of systemic amyloidosis, is brought on by long-term inflammatory conditions such as rheumatoid arthritis and persistent infections. Amyloid Transport Protein Transthyretin (ATTR)- amyloidosis is a less common familial systemic type of amyloidosis [3-5]. A more recent study has shown that the prevalence might be higher, and even more than AL.
The GI tract, especially localized GI amyloidosis, is relatively uncommon. Even when affected, only 30–60% of patients develop symptoms. The often vague and nonspecific presentation of amyloidosis contributes to diagnostic delays. Symptoms may include dysphagia, abdominal pain, gastrointestinal bleeding, constipation, diarrhea, and malabsorption [6]. Importantly, the pattern of amyloid deposition varies by amyloid type and influences clinical and endoscopic findings. In AL amyloidosis, amyloid typically deposits in the muscularis mucosae, submucosa, and muscularis propria, including vascular walls, which can manifest as polypoid lesions or thickened folds. Conversely, AA amyloidosis often involves the lamina propria, producing erosions [7,8]. These pathological differences contribute to differing clinical presentations: AL amyloidosis is more likely to cause constipation, bowel obstruction, or pseudo-obstruction, whereas AA amyloidosis more commonly results in diarrhea and malabsorption.
In this systematic review, we assess the subtypes associated with GI involvement and associated symptom prevalence.
Methods
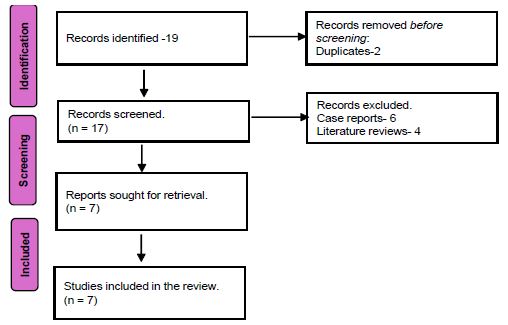
PRISMA guidelines were followed. MeSH terms including (Amyloidosis) AND (Gastrointestinal tract) were used. Cochrane, PubMed, and Google Scholar were used as search engines. 19 studies resulted, 2 were duplicates, 6 were case reports, and 4 were literature reviews, leaving 7 studies for review (Figure 1).
Results and discussion
Amyloid subtypes in GI amyloidosis
Table 1: Studies included in the review.
| Study | Author | Years | Number of patients |
|---|---|---|---|
| Gastrointestinal amyloidosis: an often-unexpected finding with systemic implications [9] | Catherine et al. | 2008-2021 | 2511 |
| Amyloidosis of the gastrointestinaltract: a 13-year, single-center, referral experience [10] | Andrew etal. | 1998 -2011 | 76- Systemic amyloidosis with dominant gastrointestinal involvement was present in 60 (79%)patients, whereas the other 16 (21%) patients had amyloidosis localized to thegastrointestinal tract |
| Clinical features and outcomes of systemic amyloidosis with gastrointestinal involvement: a single-center experience [11] | Young et al. | 1995 -2013 | 137 |
| First Nationwide Surveyof 199 Patients with Amyloid A Amyloidosis in Japan [12] | Yasuaki et al. | 2012-2014 | 199patients with AA, 132 withbiopsy-proven GI involvement, and 80 patients with GI symptoms |
| THAOS: Gastrointestinal manifestations of transthyretin amyloidosis - com- mon complications of a rare disease [13] | Jonas et al. | 2013 | 984 |
| Intestinal pseudo-obstruction in patients withamyloidosis: clinicopatho- logic differences between chemical typesof amyloid protein [14] | Tada et al. | 1993 | 16 |
| A systematic reviewof the literature on localized gastrointestinal tract amyloidosis:Presentation, management and outcomes [15] | Mariuxiet al. | 2024 | 62 |
Table 2: Subtypes of amyloids deposited.
| Amyloid Type | Frequency (approximate range across studies) |
|---|---|
| AL (LightChain) | 55–83% |
| ATTR (Transthyretin) | 4–11% |
| AA (Amyloid A) | 6–13% |
| Others | (Lesser-known subtypes like AApoAIV, ALys, Aβ2M, etc.) | 2–4% |
Table 3: Symptoms.
| Symptom | Frequency Range |
|---|---|
| Diarrhea | 32–46% (Studies 1, 3, 4) |
| Weight loss | 29–45% |
| GI bleeding (melena oroccult) | 27–36% |
| Nausea/vomiting | 29.2% |
| Early satiety, anorexia | 26–37.5% |
| Paralytic ileus / pseudo-obstruction | Rare, mostlyAA/AL cases |
Across the six studies, the most common GI amyloid types were: Catherine et al (N=2511) found AL (77.9%) to be the predominant type, followed by ATTR (11.3%) and AA (6.6%). Andrew et al. reported that 83% of systemic GI cases were AL, with rare ATTR and lysozyme amyloidosis. Young et al. showed similar trends: AL (83.3%), AA (12.5%), and ATTR (4.2%). Yasuaki et al., Jonas et al., and Tada et al. focused more on AA and ATTR, highlighting subtype-specific complications.
Clinical manifestations
Common GI symptoms across all studies: In Young et al., Diarrhea and anorexia were most common in AL. Young et al. had Intractable diarrhea in 32.2% of AA amyloidosis. Jonas et al. showed early satiety and weight loss were dominant in hereditary ATTR. Tada et al. had pseudo-obstruction linked with AA and AL types.
Histopathologic distribution
Table 4: Site involvement.
| Site | Most Commonin AL(Young et al.) | Most common,both AL and AA (in Mariuxi et al.) |
|---|---|---|
| Stomach | 55% | 42% |
| Colon | 45% | 24% |
| Rectum | 35% | 5% |
| Small bowel | 5% | 26% |
| Esophagus | 8% |
Systemic involvement and prognosis
Table 5: Extra findings from the studies.
| AL Amyloidosis | • Commonly associated with cardiac involvement
(83.5% in Catherine et al.). • Systemic presentation was dominant in 79% (Andrew et al.). • GI involvement predicted shorter survival (median 7.95 months vs 15.84 months without GI involvement; Young et al.). |
| ATTR Amyloidosis | • 100% cardiac involvement in Catherine et al. • GI symptoms more pronounced in hereditary ATTR with V30M mutations (Jonas et al.). |
| AA Amyloidosis | • Linked to chronic inflammation (e.g., rheumatoid arthritis, IBD, Castleman’s disease). • Renal and GI symptoms common (Young et al.). • GI involvement included diarrhea, melena, ileus. • Pseudo-obstruction linked to myopathy/neuropathy and differed by subtype (Tada et al.). |
People presenting with GI manifestations of amyloidosis have non-specific symptoms including nausea, vomiting, diarrhea, gastrointestinal bleeding, bowel obstruction, malabsorption, and weight loss [6]. In our study, diarrhea and weight loss were most prominent, followed by anorexia, GI bleeding, and nausea. These symptoms are nonspecific and were often preceded by empiric therapies and diagnostic tests. The nonspecific symptoms often lead to a delay in endoscopy, biopsy, and thereby diagnosis. Most of the patients had pre-existing systemic disease or other organ involvement, which suggests an early consideration of biopsy for refractory symptoms.
Any site of the GI tract can be affected, but the small bowel is most affected [16]. However, in our study, the stomach was the most prominently involved organ, followed by the colon, and then the small bowel. Further, endoscopic appearance may be highly variable with AL variants producing polyps and thickened mucosa, whereas AA counterparts produce more erosive changes with overlaps [17-19].
AL involvement was most prominent in our study, followed by ATTR and then AA. AL disease was associated with more systemic involvement and higher mortality than GI disease. This generally correlates with malignant gammopathies associated with AL, with GI involvement possibly indicating advanced disease. ATTR had GI involvement with hereditary disease, and AA with multisystem rheumatic diseases, with prominent cardiac and renal involvement, respectively. A study of 137 AL amyloidosis patients found median survival times of 7.95 months for those with GI involvement compared to 15.84 months for those without GI involvement [11].
With a sensitivity of 75% to 85%, a rectal mucosal biopsy with Congo red stain is a feasible screening alternative for patients who need endoscopy [20]. Amyloidosis is frequently identified as AL type only after AA and Transthyretin (ATTR) types have been ruled out by immunohistochemistry and hereditary kinds have been identified by genetic sequencing. Given that bone marrow involvement by SAP scintigraphy is nearly diagnostic of AL and that a specific pattern of abnormal uptake, including in the heart, by DPD scintigraphy is strongly suggestive of cardiac ATTR amyloidosis [21].
Treating the underlying condition, whether it be an autoimmune illness, infection, or cancer, is the mainstay of treatment for acquired amyloidosis. Treatment of any monoclonal gammopathy, regardless of clone burden, is indicated for AL amyloidosis with evidence of organ damage. 72.7% of patients treated with anti-Interleukin 6 receptor (IL-6) agents experienced clinical and laboratory remission after five years, compared to 40.7% of patients receiving anti-Tumor Necrosis Factor therapy (TNF), according to a retrospective study of 42 patients with AA amyloidosis caused by different rheumatic diseases. Lastly, supportive care includes treating diarrhea with octreotide, treating any infections that may arise from treatment-related immunological impairment, and providing food and vitamin supplements for individuals with malabsorption. Treatment Options for ATTR include TTR stabilizers like tafamidis and acoramidis, helping to prevent it from misfolding and forming amyloid deposits.
Conclusion
GI amyloidosis, though often underrecognized, is caused by AL, ATTR, and AA subtypes. Symptoms like diarrhea, weight loss, and GI bleeding should prompt histological evaluation using Congo red stain and subtype confirmation via proteomics. Cardiac and renal involvement is frequent in systemic cases. Recognizing and subtyping GI amyloid early is essential for improving prognosis and guiding treatment.
References
- Real de Asúa D, Costa R, Galván JM, Filigheddu MT, Trujillo D, Cadiñanos J. Systemic AA amyloidosis: epidemiology, diagnosis, and management. Clin Epidemiol. 2014; 6: 369–77.
- Sattianayagam PT, Hawkins PN, Gillmore JD. Systemic amyloidosis and the gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2009; 6: 608–17.
- Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992; 79: 1817–22.
- Real de Asua D, Costa R, Galvan JM, et al. Systemic AA amyloidosis: epidemiology, diagnosis, and management. Clin Epidemiol. 2014; 6: 369–77.
- Jadoul M, Drüeke TB. Beta2 microglobulin amyloidosis: an update 30 years later. Nephrol Dial Transplant. 2016; 31: 507–9.
- Petre S, Shah IA, Gilani N. Gastrointestinal amyloidosis: clinical features, diagnosis and therapy. Aliment Pharmacol Ther. 2008; 27: 1006–16.
- Kaiserling E, Krober S. Massive intestinal hemorrhage associated with intestinal amyloidosis: an investigation of underlying pathologic processes. Gen Diagn Pathol. 1995; 141: 147–54.
- Hirschfield GM. Amyloidosis: a clinico-pathophysiological synopsis. Semin Cell Dev Biol. 2004; 15: 39–44.
- Hagen CE, Dasari S, Theis JD, Rech KL, Dao LN, et al. Gastrointestinal amyloidosis: an often unexpected finding with systemic implications. Hum Pathol. 2023; 139: 27–36.
- Cowan AJ, Skinner M, Seldin DC, Berk JL, Lichtenstein DR, O’Hara CJ, et al. Amyloidosis of the gastrointestinal tract: a 13-year, single-center, referral experience. Haematologica. 2013; 98: 141–6.
- Lim AY, Lee JH, Jung KS, Gwag HB, Kim DH, Kim SJ, et al. Clinical features and outcomes of systemic amyloidosis with gastrointestinal involvement: a single-center experience. Korean J Intern Med. 2015; 30: 496–505.
- Okuda Y, Yamada T, Ueda M, Ando Y. First nationwide survey of 199 patients with amyloid A amyloidosis in Japan. Intern Med. 2018; 57: 3351–5.
- Wixner J, Mundayat R, Karayal ON, Anan I, Karling P, Suhr OB. THAOS investigators. THAOS: gastrointestinal manifestations of transthyretin amyloidosis—common complications of a rare disease. Orphanet J Rare Dis. 2014; 9: 61.
- Tada S, Iida M, Yao T, Kitamoto T, Yao T, Fujishima M. Intestinal pseudo-obstruction in patients with amyloidosis: clinicopathologic differences between chemical types of amyloid protein. Gut. 1993; 34: 1412–7.
- Malone MAV, Castillo DAA, Santos HT, Kaur A, Elrafei T, Steinberg L, Kumar A. A systematic review of the literature on localized gastrointestinal tract amyloidosis: presentation, management and outcomes. Eur J Haematol. 2024; 113: 400–15.
- Syed U, Ching Companioni RA, Alkhawam H, et al. Amyloidosis of the gastrointestinal tract and the liver: clinical context, diagnosis and management. Eur J Gastroenterol Hepatol. 2016; 28: 1109–21.
- Hokama A, Kishimoto K, Nakamoto M, Kinjo N, Kinjo F, Fujita J. Endoscopic and histopathological features of gastrointestinal amyloidosis. World J Gastrointest Endosc. 2011; 3: 157–61.
- Tada S, Iida M, Yao T, et al. Endoscopic features in amyloidosis of the small intestine: clinical and morphologic differences between chemical types of amyloid protein. Gastrointest Endosc. 1994; 40: 45–50.
- Alcalde-Vargas A, Leo-Carnerero E, Rojas-Mercedes N, et al. Correlation between location of amyloid deposits and endoscopic and clinical manifestations in symptomatic gastrointestinal amyloidosis. Rev Esp Enferm Dig. 2015; 107: 49–51.
- Hachulla E, Grateau G. Diagnostic tools for amyloidosis. Joint Bone Spine. 2002; 69: 538–45.
- Gillmore JD, Wechalekar A, Bird J, et al. Guidelines on the diagnosis and investigation of AL amyloidosis. Br J Haematol. 2015; 168: 207–18.
- Okuda Y, Ohnishi M, Matoba K, et al. Comparison of the clinical utility of tocilizumab and anti-TNF therapy in AA amyloidosis complicating rheumatic diseases. Mod Rheumatol. 2014; 24: 137–43.
- Shin JK, Jung YH, Bae MN, et al. Successful treatment of protein-losing enteropathy due to AA amyloidosis with octreotide in a patient with rheumatoid arthritis. Mod Rheumatol. 2013; 23: 406–11.