
Journal of Gastroenterology Research and Practice
Research Article - Open Access, Volume 4
Mitochondrial proteins and colorectal cancer risk: A mendelian randomization study
Donglei Wu1†, Dafeng Tong2†, Ming Gao3*, Xin Zhang2*, Xuguang Hu2*
1Department of Gastrointestinal Oncology, First Affiliated Hospital of China Medical University, Shenyang, China.
2Department of General Surgery, Changhai Hospital, Navy Military Medical University, Shanghai, China.
3Department of Gastroenterology, Dalian Municipal Central Hospital, Dalian, China.
†These Authors Contributed Equally to this Work.
*Corresponding Author : Xuguang Hu1; Xin Zhang2; Ming Gao3
1Department of General Surgery, Changhai Hospital, 168
Changhai Road, Shanghai 200433, China.
Email: drxuguanghu@163.com
2Department of General Surgery, Changhai Hospital, 168
Changhai Road, Shanghai 200433, China.
Email: zhangxin80@smmu.edu.cn
3Department of Gastroenterology, Dalian Municipal
Central Hospital, Dalian, China.
Email: gao_1860@163.com
Received : Apr 10, 2024
Accepted : Apr 29, 2024
Published : May 06, 2024
Archived : www.jjgastro.com
Copyright : © Hu X, Zhang X, Gao M (2024).
Abstract
Background: Mitochondrial dysfunction has been implicated in Colorectal Cancer (CRC) development. However, evidence for a causal relationship between mitochondrial proteins and CRC risk is lacking. This study aimed to explore the causal associations between mitochondrial proteins and CRC risk using a two-sample Mendelian Randomization (MR) approach.
Methods: Summary statistics data on CRC were obtained from the FinnGen consortium (n=6,509 cases, 287,137 controls). Data on genetic variants associated with 66 mitochondrial proteins were extracted from the Human Plasma Proteome Atlas (n=3,301 healthy individuals). The inverse-variance weighted method was applied as the primary analysis, with sensitivity analyses using MREgger regression, weighted median estimation, and MR-PRESSO to assess pleiotropy and outliers.
Results: Three mitochondrial proteins displayed significant causal associations with CRC risk. Increased cytochrome c oxidase subunit 8A levels were associated with increased CRC risk (OR 1.05, 95% CI 1.00-1.11, P=0.037). Decreased mitochondrial import inner membrane translocase subunit TIM14 (OR 0.86, 95% CI 0.77-0.96, P=0.010) and decreased oligoribonuclease levels (OR 0.90, 95% CI 0.83-0.98, P=0.022) were associated with reduced CRC risk. Sensitivity analyses indicated these results were robust to pleiotropy. The reverse MR analysis found no evidence for reverse causation.
Conclusion: Our MR study provides evidence that altered levels of specific mitochondrial proteins may causally influence CRC risk. Further research is warranted to elucidate the mechanisms underlying these associations. Our findings highlight potential targets for CRC prevention and treatment.
Keywords: Colorectal cancer; Mendelian randomization; Mitochondria; Genetic; Human.
Abbreviations: CRC: Colorectal Cancer; MR: Mendelian Randomization; IV: Instrumental Variable; IVW: Inverse Variance Weighted; LD: Linkage Disequilibrium; SNP: Single Nucleotide Polymorphism; OR: Odds Ratio; CI: Confidence Interval; GWAS: Genome Wide Association Study.
Citation: Wu D, Tong D, Gao M, Zhang X, Hu X. Mitochondrial proteins and colorectal cancer risk: A mendelian randomization study. J Gastroenterol Res Pract. 2024; 4(4): 1197.
Introduction
Colorectal Cancer (CRC) is the third most common cancer worldwide and is a major global health burden, as of January 2023, the cumulative number of confirmed cases is estimated to have exceeded 661 million, with a corresponding total death toll surpassing 6.7 million [1,2]. Extensive investigation into the pathogenesis of colorectal cancer has revealed a strong correlation between its occurrence and lifestyle factors, such as smoking and excessive alcohol consumption, which heighten its vulnerability [3]. Furthermore, despite the incomplete comprehension of the precise involvement of mitochondria in cancer development, multiple lines of evidence have implicated mitochondrial dysfunction in its development [4-6].
In the intricate landscape of cellular physiology, mitochondria emerge not merely as power-generating entities but as pivotal regulators of various fundamental processes. Mitochondria play diverse roles in the regulation of energy metabolism, signaling, apoptosis, and other essential cellular processes [7,8]. However, mitochondrial aberrations can upset this delicate equilibrium, opening a Pandora’s box of cellular malfunctions that potentially pave the way for tumorigenesis [9-11].
The intricate pathways and protein dynamics governing mitochondrial functions provide a vivid illustration of cellular complexity. It’s no surprise that aberrations in these processes can lead to pathological conditions, including CRC. For example, Cytochrome C Oxidase Subunit 8A (COX8A), an essential cog in the mitochondrial machinery, serves as the core subunit of complex IV in the electron transport chain [12]. This complex, responsible for the catalytic reduction of oxygen to water, is vital for the proton gradient required for ATP synthesis. Therefore, COX8A is intrinsically tied to the cell’s ATP production capacity [12]. When COX8A or its associated components are dysfunctional, cells may experience energy deprivation, compelling them to undergo metabolic reprogramming [13]. Such metabolic adaptations in cancer cells can promote the Warburg effect, where cells preferentially rely on glycolysis for energy, even under normoxic conditions. This switch provides cancer cells with both the rapid ATP generation and the metabolic intermediates necessary for biomass synthesis. Furthermore, the mitochondrial protein import machinery is integral to maintaining the organelle’s functional integrity. TIM14, part of the inner membrane translocase system, plays a crucial role in chaperoning cytosolic proteins into the mitochondrial matrix [14]. This translocation process is essential since many mitochondrial proteins are encoded by nuclear genes, synthesized in the cytosol, and subsequently imported into mitochondria. Disruptions in TIM14 or associated components can impede this process, leading to mitochondrial dysfunction due to the absence of vital proteins. Such impairments can result in an energy deficit, oxidative stress, and compromised apoptotic pathways, all of which can create a conducive environment for tumorigenesis [14].
Although these observational findings suggest that altered mitochondrial proteins contribute to CRC, evidence of a causal relationship is lacking. Establishing causality is critical, as it would better validate mitochondrial proteins as potential drug targets or biomarkers. Mendelian Randomization (MR) is a method that utilizes genetic variants associated with an exposure of interest as proxies to infer causality in an unbiased manner [15,16]. We performed a two-sample MR study to investigate the causal association between mitochondrial protein levels and CRC risk.
Material and methods
Design of the study
The study design is illustrated in Figure 1. This MR approach was implemented to minimize the potential impact of bias on the results. In this study, the MR had to adhere to three critical assumptions: (1) the Instrumental Variables (IVs) must demonstrate a significant association with proteins linked to mitochondria [17], (2) the IVs should be independent of other potential confounding factors that influence proteins associated with both mitochondria and CRC [17], and (3) the IVs should be independent of the outcome [18].
Sources of data
Table 1 summarizes the outcomes and exposures employed. Subsequently, Genome-Wide Association Study (GWAS) summary data for CRC were obtained from the FinnGen Consortium (https://www.r9.finngen.fi/, accessed on 15 August 2023). All participants were of European ancestry and provided informed consent for the FinnGen research project, which combines genetic disease endpoint data from the Finnish Biobank and Finnish Health Registry [19]. A total of 6,509 CRC cases were identified, along with 287,137 control subjects, with the inclusion of 20,167,784 Single Nucleotide Polymorphisms (SNPs). CRC was defined using the C3 code of the International Classification of Diseases-Tenth Revision (ICD-10). For comprehensive details on the participants’ information, genotyping, imputation, and quality control procedures, please refer to the FinnGen website (https://finngen.gitbook.io/documentation/). The analysis of SNPs associated with proteins related to mitochondria was conducted using summary statistics obtained from the publicly accessible Human Plasma Proteome Atlas. This atlas comprises data on 3,622 plasma proteins derived from 3,301 healthy participants who underwent genetic assessment in the INTERVAL study [20]. These data establish a connection between genetic factors and protein-based phenotypes, thereby offering potential targets for specific diseases. Following natural log transformation and adjustment for age, sex, and duration between blood draw and linear regression processing, protein concentrations were determined [20]. As our study utilized previously published research and publicly available databases, there was no requirement for additional ethical approval or informed consent.
Instrumental variable selection
IVs from the study were subject to the following quality control measures: (1) To obtain a more comprehensive result, a P-value threshold of less than 5.0×10-6 was selected to avoid too few SNPs [21]. (2) Linkage Disequilibrium (LD) represents a scenario where the inherited frequency of two genes at distinct seats within a population significantly exceeds the inherited frequency expected at random [22]. To mitigate LD, we set the imbalance threshold to R²<0.001 and the maximum distance to 10,000 kb. (3) The exclusion criteria encompassed palindromic SNPs, which means that if the alleles are A and T or C and G, then the same alleles will appear on the plus and minus chains. In the reverse MR analysis, several quality control measures were implemented to identify eligible IVs that fulfilled the three above-mentioned MR assumptions (Supplemental Table S1) [21]. Notably, a similar threshold of P<5×10-6 was employed for IV selection (Supplemental Table S2).
Data analysis
First, the Inverse Variance Weighted (IVW) approach was used as the primary approach to evaluate the relationships between proteins associated with mitochondria and CRC. After extracting the association estimates linking the instruments and outcomes, as well as harmonizing the directional orientation of these estimates as effect alleles, we applied the Wald estimator in our computation of MR estimates for each instrument, which allowed us to derive causal effect estimates [23,24]. The findings of the IVW method can be considered reliable if every SNP fulfills the MR assumptions without horizontal pleiotropy [25]. Further sensitivity analyses were employed to substantiate the robustness of the results, including the weightedmedian method, MR-Egger regression, MR-PRESSO test, and leave-one-out analyses. Weighted median estimators generate reliable causal effects if less than half of the contributing information is sourced from invalid IVs [26]. In MR-Egger regression, an intercept term P-value <0.05 is deemed statistically significant and serves as an indicator of directional pleiotropy [27]. The MR-PRESSO test was used to assess pleiotropic biases and remove outliers to correct for pleiotropic effects [28]. Finally, we performed reverse MR analysis to explore the reverse causal relationship between CRC and the proteins associated with 66 mitochondria.
F-statistics were employed to evaluate the strength of the IVs, which examined whether the effect estimates of the causal associations were affected by weak instrument bias [29]. The F-statistic was used to measure the strength of the IVs. We selected SNPs with an F-statistic >10 as IVs to satisfy a strong association with exposure [30]. When the number of SNPs was less than four, the analysis was restricted to the IVW method. Unless stated otherwise, all statistical analyses were performed using R software (version 4.1.1), with the study utilizing the ‘Two Sample MR’ and ‘MRPRESSO’ R packages. Odds Ratios (ORs) and 95% Confidence Intervals (CIs) were calculated to evaluate the association between mitochondria-associated proteins and CRC.
Results
We explored the association between 66 traits of mitochondria-associated proteins and CRC. The F-statistic of the selected IVs was >10, suggesting that there was no weakness in IVs in the above association. Among the 66 traits, three mitochondrial proteins may have increased the risk of CRC in the IVW method (Figure 2). More details are shown in Figure 3, where there was a positive association between cytochrome c oxidase subunit 8A, mitochondrial levels (COX8A.10497.242.3), and CRC risk (OR, 1.05; 95% CI, 1.00-1.11; P=0.037) in the IVW approach. However, in other methods, such as the weighted median and MR PRESSO, this causal association was consistent with the critical statistical P-value (Figure 3). Moreover, the IVW method showed that other two traits may decrease the risk of CRC: mitochondrial import inner membrane translocase subunit TIM14 levels (DNAJC19.4545.53.3) (OR, 0.86; 95% CI, 0.77-0.96; P=0.010), and mitochondrial oligoribonuclease levels (REXO2.13590.1.3) (OR, 0.90; 95% CI, 0.83-0.98; P=0.022). Similar associations were observed using the MR-PRESSO approach (Figure 3). In addition, the intercepts of the MR-Egger results suggested that these causalities were not influenced by potential pleiotropy (all P-values >0.05), and MR PRESSO did not find outlier SNPs among the selected IVs for these traits. Additional information on the association of mitochondrial proteins with CRC is presented in Supplementary Table 3 and Supplementary Table 4. In the reverse MR analysis, we did not find any specific mitochondrial proteins associated with CRC, suggesting that reverse causation is unlikely to be the cause (Supplementary Table 5).
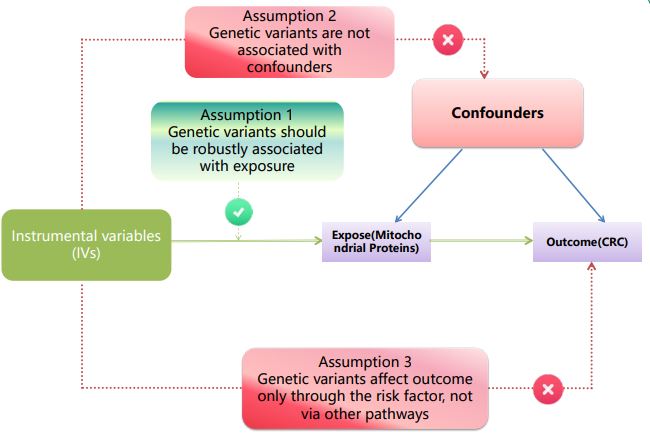
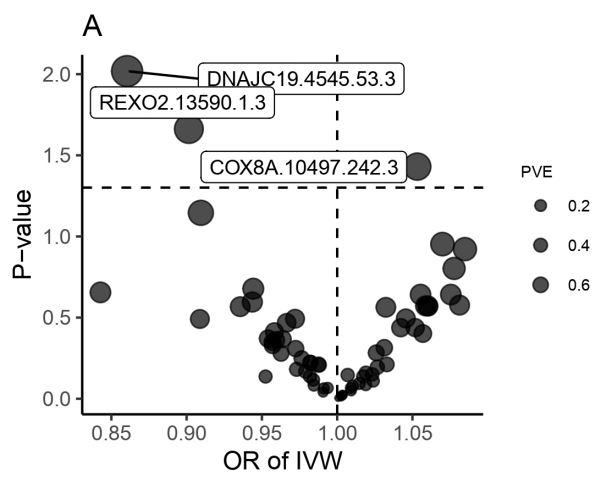
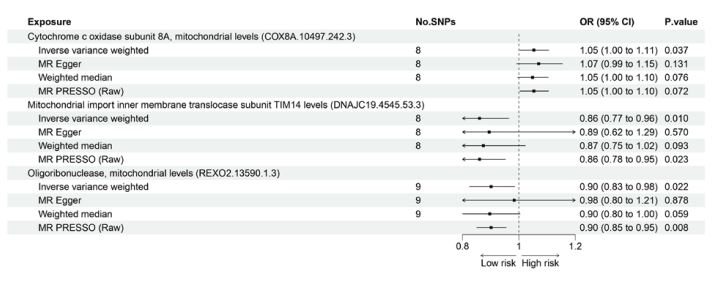
Table 1:Details of the genome-wide association studies and datasets used in our analyses.
| Exposure or outcome | Sample size | Ancestry | Link | PMID |
|---|---|---|---|---|
|
Genomic atlas of the
human plasma proteome |
3,301 participants | European | https://www.ebi.ac.uk/gwas/publications/29875488 | 29875488 |
| Colorectal cancer |
6,509 cases and 287,137 controls |
European |
https://storage.googleapis.com/finngen-public-data-r9/summary_ stats/finngen_R9_C3_COLORECTAL_EXALLC.gz |
/ |
Discussion
In this two-sample MR study, we found evidence of a causal association between mitochondrial protein levels and CRC risk. Specifically, increased levels of cytochrome c oxidase subunit 8A are associated with an increased CRC risk. In contrast, decreased levels of mitochondrial import inner membrane translocase subunit TIM14 and oligoribonuclease were linked to reduced CRC risk.
The cytochrome c oxidase complex plays a critical role in mitochondrial oxidative phosphorylation, and disruption of its function can promote cancer development [31]. This may reflect the increased turnover or instability of cytochrome c oxidase subunits in the tumor microenvironment. Dysregulation of cytochrome c oxidase subunits has been reported in multiple cancer types, including lung, breast, and prostate cancer [32- 35]. The decreased expression of cytochrome c oxidase subunits may cause energy stress and metabolic reprogramming in cancer cells [36]. Cancer cells adapt by increasing glycolysis and pentose phosphate pathway activity, providing intermediates for the anabolic processes required for rapid proliferation [37]. Several mechanisms may be attributed to the link between altered cytochrome c oxidase subunit 8A levels and CRC risk. Subunit 8A expression may be disrupted by abnormalities in gene regulation or mitochondrial biogenesis pathways. Decreased import or defective incorporation of subunit 8A into the cytochrome c oxidase complex can impair its catalytic activity and electron transport function, thereby promoting oxidative stress [38]. The oxidative damage and energy deficits arising from cytochrome c oxidase dysfunction may generate further mitochondrial abnormalities and mutations [39]. This can initiate a deleterious positive feedback loop, further exacerbating mitochondrial dysfunction during CRC development.
In addition to the direct effects on energy metabolism, the disruption of cytochrome c oxidase subunit levels may also affect cancer-related signaling pathways. Cytochrome c oxidase subunits interact with proteins involved in cell cycle regulation, apoptosis, hypoxia responses, and metastasis [40,41]. The altered subunit composition of the cytochrome c oxidase complex can disrupt the proper assembly and functioning of these signaling networks. Additional effects may arise from increased electron leakage and reactive oxygen species generation due to impaired cytochrome c oxidase activity. Further research is warranted to elucidate the mechanisms through which altered levels of cytochrome c oxidase subunit 8A promote CRC tumorigenesis. This may reveal new therapeutic targets aimed at restoring cytochrome c oxidase function and normalizing metabolism in CRC.
We also found evidence that decreased levels of mitochondrial import inner membrane translocase subunit TIM14 are causally associated with a reduced CRC risk. TIM14 is essential for the translocation of nuclear-encoded proteins to the mitochondrial matrix [14]. It acts as a receptor on the inner membrane to facilitate the pulling of precursor proteins across the aqueous intermembrane space. TIM14 downregulation impairs the import of proteins required for mitochondrial activity and cellular proliferation [14]. Our results indicate that decreased TIM14 levels may suppress CRC development, possibly by impairing mitochondrial protein import. Thus, reduced TIM14 function may contribute to the development of CRC through multiple mechanisms. Defective mitochondrial protein import leads to the accumulation of unfolded proteins in the intermembrane space, causing proteotoxic stress [42]. Impaired import of proteins involved in mitochondrial gene expression, fission/fusion, and quality control can disrupt organelle homeostasis. The lack of important matrix enzymes, such as those required for the tricarboxylic acid cycle or oxidative phosphorylation, may deplete the metabolites needed for anabolic processes. Furthermore, reduced mitochondrial protein import prevents the maturation of apoptosis-regulating factors, such as HtrA2/Omi, thereby inhibiting cell death [43]. Additional effects may arise from the impaired import of cytosolic pro-oncogenic signaling proteins that translocate to the mitochondria. Further studies are required to validate the functional effects of altered TIM14 expression on mitochondrial activity and CRC pathogenesis.
Finally, we identified a causal relationship between low oligoribonuclease levels and reduced CRC risk. As a 3’-exoribonuclease, oligoribonuclease degrades small RNAs and regulates metabolic homeostasis [44]. It is localized in the mitochondrial matrix, where it mediates the degradation of mitochondriaencoded RNAs [44]. Oligoribonucleases have also been linked to the regulation of signaling pathways involved in proliferation, metastasis, and apoptosis. Our findings suggest that oligoribonuclease activity is required for CRC growth and survival. There are several mechanisms by which oligoribonucleases hinder CRC development. Impaired mitochondrial RNA degradation may disrupt organelle gene expression, causing respiratory chain deficits [45]. Accumulation of mitochondrial small non-coding RNAs can dysregulate transcription or epigenetic programming, thereby influencing cancer phenotypes [46,47]. Oligoribonucleases may also directly target cytoplasmic RNAs involved in oncogenesis [48].
Conclusion
Overall, our study provides novel evidence that mitochondrial proteins play a causal role in CRC development. Although promising, MR studies have limitations, including the potential pleiotropic effects of genetic variants. The use of multiple sensitivity analyses mitigated this concern. Population stratification can also confound the MR results; however, this was minimized by using extensive genotype data to restrict our analysis to individuals of European ancestry. Residual confounding is another potential issue, as our analysis focused specifically on mitochondrial protein levels, whereas many other factors contribute to CRC risk. Additional MR studies on other CRC risk factors could help build a more comprehensive map of the causal mechanisms. Furthermore, although MR can be used to infer causality, it cannot determine the specific biological pathways involved. Future studies should build on our findings by performing proteomic analyses of CRC patient samples and in vitro experiments that manipulate mitochondrial protein levels in CRC cell lines and animal models.
In summary, our MR analysis provides novel evidence for a causal association between mitochondrial protein levels and CRC risk. These findings implicate cytochrome c oxidase subunit 8A, mitochondrial import inner membrane translocase subunit TIM14, and oligoribonuclease as potential mediators influencing colorectal tumorigenesis through their effects on mitochondrial pathways. Additional studies are warranted to confirm the functional relevance of these proteins in CRC pathogenesis. Elucidating the mechanisms linking the altered expression of these mitochondrial proteins to cancer-associated pathways could reveal new prognostic biomarkers and therapeutic targets for CRC. Our study highlights the potential application of genetic approaches to gain insights into the causal mechanisms of CRC development. Further research building on these results may ultimately translate into improved prevention, diagnosis, and treatment strategies to reduce the burden of this major malignancy.
Declarations
Conflicts of interest: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethics approval: All studies included in the GWASs had been approved by relevant ethical review committees, and all participants had provided written informed consent. The current study only used summary-level data that were publicly available; thus, no additional ethical review was required for this study.
Informed consent: Not applicable.
Acknowledgments: The author expresses profound gratitude to the researchers and participants of the initial genomewide association studies for their meticulous collection and stewardship of extensive data sets. Additionally, appreciation is extended to all individuals who played an active role in the present investigation
Funding: No funding.
Data availability statement: All relevant data has been presented in the manuscript and further inquiry can be directed to the corresponding author.
References
- Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011; 61: 212-236.
- Nga NTV, Xuan VN, Trong VA, Thao PH, Doanh DC. Perceived Barriers and Intentions to Receive COVID-19 Vaccines: Psychological Distress as a Moderator. Vaccines (Basel). 2023; 11.
- Wu C, Wang M, Shi, H. Cholesterol Promotes Colorectal Cancer Growth by Activating the PI3K/AKT Pathway. J Oncol 2022; 1515416.
- Lin PC, et al. Expression of beta-F1-ATPase and mitochondrial transcription factor A and the change in mitochondrial DNA content in colorectal cancer: Clinical data analysis and evidence from an in vitro study. Int J Colorectal Dis. 2008; 23: 1223-1232.
- Zhang, W. et al. Multi-omics analyses of human colorectal cancer revealed three mitochondrial genes potentially associated with poor outcomes of patients. J Transl Med. 2011; 19: 273.
- Yun CW, Han YS, Lee SH. PGC-1alpha Controls Mitochondrial Biogenesis in Drug-Resistant Colorectal Cancer Cells by Regulating Endoplasmic Reticulum Stress. Int J Mol Sci. 2019; 20.
- Tak H, et al. T-cell-restricted intracellular antigen 1 facilitates mitochondrial fragmentation by enhancing the expression of mitochondrial fission factor. Cell Death Differ. 2017; 24: 49-58.
- Liu Y, et al. An Overview: The Diversified Role of Mitochondria in Cancer Metabolism. Int J Biol Sci. 2023; 19: 897-915.
- Donath S, et al. Apoptosis repressor with caspase recruitment domain is required for cardioprotection in response to biomechanical and ischemic stress. Circulation. 2006; 113: 1203-1212.
- Chen K, et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin Cancer Biol. 2022; 83: 556-569.
- Liu Y, et al. Emerging phagocytosis checkpoints in cancer immunotherapy. Signal Transduct Target Ther. 2023; 8: 104.
- Zhao N, et al. After Treatment with Methylene Blue is Effective against Delayed Encephalopathy after Acute Carbon Monoxide Poisoning. Basic Clin Pharmacol Toxicol. 2018; 122: 470-480.
- Figueiredo-Pereira, C Dias-Pedroso, D Soares NL, Vieira HLA. CO-mediated cytoprotection is dependent on cell metabolism modulation. Redox Biol. 2020; 32: 101470.
- Mokranjac D, Bourenkov G, Hell K, Neupert W, Groll M. Structure and function of Tim14 and Tim16, the J and J-like components of the mitochondrial protein import motor. EMBO J. 2006; 25: 4675-4685.
- Eriksson J, et al. Causal relationship between obesity and serum testosterone status in men: A bi-directional mendelian randomization analysis. PLoS One. 2017; 12: e0176277.
- Markozannes G, et al. Systematic review of Mendelian randomization studies on risk of cancer. BMC Med. 2022; 20: 41.
- Davies NM, Holmes MV, Davey Smith G. Reading Mendelian randomisation studies: A guide, glossary, and checklist for clinicians. BMJ. 2018; 362: k601.
- Davey Smith G, Hemani G. Mendelian randomization: Genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014; 23: R89-98.
- Chen Y, et al. Tea consumption and risk of lower respiratory tract infections: A two-sample mendelian randomization study. Eur J Nutr. 2023; 62: 385-393.
- Sun BB, et al. Genomic atlas of the human plasma proteome. Nature. 2018; 558: 73-79.
- Liu B, et al. Associations of the circulating levels of cytokines with risk of amyotrophic lateral sclerosis: A Mendelian randomization study. BMC Med. 2023; 21: 39.
- Roze D. Causes and consequences of linkage disequilibrium among transposable elements within eukaryotic genomes. Genetics. 2023; 224.
- Lee CH, Cook S, Lee JS, Han B. Comparison of Two Meta-Analysis Methods: Inverse-Variance-Weighted Average and Weighted Sum of Z-Scores. Genomics Inform. 2016; 14: 173-180.
- Hartwig FP, Borges MC, Horta BL, Bowden J, Davey Smith G. Inflammatory Biomarkers and Risk of Schizophrenia: A 2-Sample Mendelian Randomization Study. JAMA Psychiatry. 2017; 74: 1226-1233.
- Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013; 37: 658-665.
- Bowden J, Davey Smith G, Haycock PC, Burgess, S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol. 2016; 40: 304-314.
- Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015; 44: 512-525.
- Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018; 50: 693-698.
- Palmer TM, et al. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res. 2012; 21: 223-242.
- Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for Mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011; 40: 740-752.
- Shapiguzov A, et al. Arabidopsis RCD1 coordinates chloroplast and mitochondrial functions through interaction with ANAC transcription factors. Elife 8. 2019.
- Zhou W, et al. Peripheral blood mitochondrial DNA copy number is associated with prostate cancer risk and tumor burden. PLoS One. 2014; 9: e109470.
- Lee HC, Yin PH, Chi CW, Wei YH. Increase in mitochondrial mass in human fibroblasts under oxidative stress and during replicative cell senescence. J Biomed Sci. 2022; 9: 517-526.
- Ye Z, et al. Comprehensive Analysis of Alteration Landscape and Its Clinical Significance of Mitochondrial Energy Metabolism Pathway-Related Genes in Lung Cancers. Oxid Med Cell Longev. 2021; 9259297.
- Chen K, et al. Advances in the Prevention and Treatment of Obesity-Driven Effects in Breast Cancers. Front Oncol. 2022; 12: 820968.
- Caron-Debarle M, et al. Adipose tissue as a target of HIV-1 antiretroviral drugs. Potential consequences on metabolic regulations. Curr Pharm Des. 2010; 16: 3352-3360.
- Olivier-Van Stichelen S, et al. O-GlcNAcylation stabilizes betacatenin through direct competition with phosphorylation at threonine 41. 2014; 28: 3325-3338.
- Urbaniak B, Nowicki P, Sikorska D, Samborski W, Kokot ZJ. The feature selection approach for evaluation of potential rheumatoid arthritis markers using MALDI-TOF datasets. Anal Biochem. 2017; 525: 29-37.
- Chandra D, Singh KK. Genetic insights into OXPHOS defect and its role in cancer. Biochim Biophys Acta. 2011; 1807: 620-625.
- Lee HC, Wei YH. Mitochondrial role in life and death of the cell. J Biomed Sci. 2000; 7: 2-15.
- Huttemann M, Lee I, Samavati L, Yu H, Doan JW. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim Biophys Acta. 2007; 1773: 1701-1720.
- Wrobel L, et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature. 2015; 524: 485-488.
- Burri L, et al. Mature DIABLO/Smac is produced by the IMP protease complex on the mitochondrial inner membrane. Mol Biol Cell. 2005; 16: 2926-2933.
- Lee CW, et al. Structural basis of small RNA hydrolysis by oligoribonuclease (CpsORN) from Colwellia psychrerythraea strain 34H. Sci Rep. 2019; 9: 2649.
- Metodiev MD, et al. Recessive Mutations in TRMT10C Cause Defects in Mitochondrial RNA Processing and Multiple Respiratory Chain Deficiencies. Am J Hum Genet. 2016; 99: 246.
- Morita S, et al. Genome-wide analysis of DNA methylation and expression of microRNAs in breast cancer cells. Int J Mol Sci. 2012; 13: 8259-8272.
- Burzio VA, et al. Expression of a family of noncoding mitochondrial RNAs distinguishes normal from cancer cells. Proc Natl Acad Sci USA. 2009; 106: 9430-9434.
- Qian Y, et al. Upregulation of the long noncoding RNA UCA1 affects the proliferation, invasion, and survival of hypopharyngeal carcinoma. Mol Cancer. 2017; 16: 68.