
Journal of Gastroenterology Research and Practice
Review Article - Open Access, Volume 3
Is the epithelial-mesenchymal transition an ally or an enemy of cancer?
Juarez Lopez Ana L1,2,3; Maldonado Bernal Carmen2; Sánchez Zauco Norma A2,3*
1Bachelor of Medicine, Benemérita Autonomous University of Puebla, México.
2Immunology and Proteomics Research Unit, Children’s Hospital of Mexico “Federico Gomez”, México City, México.
3Division of Diagnostic and Treatment Auxiliaries UMAE, Pediatrics Hospital “Dr. Silvestre Frenk Freund”, National Medical Center Siglo XXI, IMSS, 06720, Mexico City, Mexico.
*Corresponding Author : Sánchez Zauco Norma A
Immunology and Proteomics Research Unit, Children’s
Hospital of Mexico “Federico Gomez”, México City,
México.
Email: angi_sanz5@yahoo.com.mx
Received : Nov 15, 2023
Accepted : Dec 14, 2023
Published : Dec 21, 2023
Archived : www.jjgastro.com
Copyright : © Sánchez Zauco NA (2023).
Abstract
The cancer continues to be a leading cause of mortality in the world, the increment number of the cases has been correlated with the mechanisms that the tumoral cells have for growth and metastasis, there are some processes for the survival and resistance of cancer cells including epithelial-mesenchymal transition, metastasis, drug resistance and angiogenesis. The EMT is a biological process that allows cells of epithelial lineage undergo to a conversion to mesenchymal cell phenotype resulting in cells in intermediate states that have both, epithelial and mesenchymal characteristics. The ETM process induced pluripotent stem cell reprogramming and confers to the cancer cells tumor-initiating and metastatic potential. The development of metastatic process involves some proteins like E-cadherin, HLA-E, SPARC, TNS-4 and FN-1, each protein has an important role as check point of the metastasis and allow immune evasion and grows the seconds tumor.
Keywords: ETM; Metastasis; Immune evasion; E-cadherin; HLAE; SPARC; TNS-4; FN-1.
Abbreviations: EMT: Epithelial-Mesenchymal Transition; PMN: Premetastatic Niche; TME: Tumoral microenvironment; CAFs: Cancer Associated Fibroblasts, TAMs: Immune Cells Like Tumor-Associated Macrophages; CTCs: Circulating tumor cells; BMDCs: Bone Marrow-Derived Cells; MHC: Major Histocompatibility Complex; CTL: Cytotoxic T lymphocyte; TILs: Tumor-Infiltrating Lymphocytes; NK: Natural Killer.
Citation: Juarez Lopez AL, Maldonado BC, Sánchez Zauco NA. Is the epithelial-mesenchymal transition an ally or an enemy of cancer?. J Gastroenterol Res Pract. 2023; 3(10): 1174.
Introduction
Despite the continuous deployment of new diagnostic and treatment strategies, cancer continues to be a leading cause of mortality. Based on the GLOBOCAN 2020 dates, there were almost 10.0 million cancer deaths, 19,292,789 new cases in the world in 2020, and forecast almost 28.9 million new cases in 2040 [1].
The increment number of the cases has been correlated with the mechanisms that the tumoral cells have for growth and metastasis [2]. Recently evidence, have been identified common features involves in cancer development that have been dubbed hallmarks of cancer”. These hallmarks are characteristics of the complex biological capabilities acquired during the development of tumors [3] and one of these hallmarks is the evasion of the immune response [4].
Tumor cells use diverse steps to avoid effectively the immune system [5]. One of these pathways is correlated with the inhibitory and stimulatory immune checkpoint molecules, the expression of these molecules in diverse tumor types (like melanoma, renal, ovarian and lung carcinoma [6-9] has been related to important processes for the survival and resistance of cancer cells including epithelial-mesenchymal transition, metastasis, drug resistance and angiogenesis [10].
Diverse studies have shown that epithelial-mesenchymal transition (EMT) is associated with a variety of immunosuppressive cells, and the expression of immune checkpoints, such as programmed cell death-ligand 1, in several cancer types [11- 14]. Also, the epithelial-mesenchymal transition (EMT) tumor cells have a stretch crosstalk with the immune cells providing EMT cells promoting immune exclusion [15].
The EMT is a cellular program that confers to the cancer cells tumor-initiating and metastatic potential [16]. Furthermore epithelial-mesenchymal transition (EMT) [17] increased the success of immunotherapy [18,19].
Some proteins like as HLA-E and SPARC (secreted protein acidic and cysteine rich) recently have correlated with the immune evasion of tumoral cells [20,21], also proteins like Ecadherin have also been related to the EMT in different types of cancer [22,75,81,88]. Therefore, knowing about the mechanisms and the molecules implicated in the immune evasion and the EMT process are essential to ensure the most effective therapy, reduce the damaging effects of treatment, and direct the therapy to specific targets. In this article, we will briefly present an overview of how cancer evades immunity, the relation between the EMT and immune evasion, and how some proteins like E-cadherin, HLA-E, SPARC, TNS-4 and FN-1 have correlated with these processes.
Tumor immune evasion
The immune system is shaped by a variety of cells and molecular mechanisms [23] that are equilibrate for the recognition, elimination and memorize of harmful agents such as virus, bacteria, allergenic substance and even cellular alterations such as tumoral cells in the organism. However, this synchronized process of recognition to memorize has been altered by the cancer cells using the host immunity mechanisms to grow and disseminate in the organism even if the host has a functional immune system. This cancer activity had been studied for decades and receive the name of immunoediting [24,25].
The process of immunoediting is conformed for three stages: an initial one, knowing like -elimination- where the immune system can recognize and eliminate tumoral cells by innate and adaptive immune cells [26], the second one is equilibrium, where the malignant cells that could be destroyed remain and develop greater adaptation of the tumor-cells consequently for the pressure exercised by the immune system on the genetically and unstable highly heterogeneous cells in the tumor mass [27]. The final stage is escape or evasion, this happens when the selected cancer cells lack control thereby grow and disseminate in an immunocompetent environment [28].
Therefore tumors exploit several immunological processes such as the use of regulatory T cells (Tregs); CD4+CD25+FoxP3+ for the immune suppression [29]. Diverse studies have shown that tumor-derived Tregs have comparatively higher suppressive activity than naturally occurring Tregs [30,31] this is probably caused using multiple suppressive mechanisms to reduce tumor-associated antigen (TAA)-specific T-cell immunity [32]. The defective antigen presentation is another mechanism used by the tumoral cells and this occurs because of the affectation of the major histocompatibility complex (MHC) secondary to the down modulating antigen processing machinery. Therefore, the expression of tumor antigen is downregulated and has correlated with a cytotoxic T lymphocyte (CTL) with low capacity for recognition of target antigens on the tumor cells [33].
Another fundamental mechanism by which tumors can evade immune surveillance is by crippling CTL functionality via the production of immune-suppressive cytokines, especially by immune cells and epithelial cells [34]. For example, TFG-α which is the principal mediator of this activity has correlated with the inhibit maturation of dendritic cells (DC’s) and cytokines like tumor necrosis factor (TNF-α), IL-1, IL-6, colony-stimulating factor (CSF)-1, IL-8, IL-10 can also significantly contribute to cancer growth [34,36,37] also studies had demonstrated that VEGF produced by tumors, inhibits the differentiation of progenitors into DC [38]. Pericyte are specialized cells that are in intimated contact with the basement membrane of capillaries, they coordinate intercellular signaling with the other components of the blood vessel [39], this cells also are implicated indirectly in tumor growth and metastasis tumor by diverse mechanisms for example the vasculature hyperstimulated by VEGF often has reduced pericyte coverage, its decrease facilitating metastatic spread [40].
Another component is the immune deviation produced by shifting the balance from Th1 to Th2 in a TGF-β- and IL-10-dependent manner [41], furthermore the expression of inhibitory molecules like programmed cell death (PD)-L1/B7H1 on tumor cells also has been associated with deletion or energy on tumor-reactive cells [42,43].
Epithelial-mesenchymal transition and immunity
The Epithelial-Mesenchymal Transition (EMT) is a biological process that allows cells of epithelial lineage undergo to a conversion to mesenchymal cell phenotype resulting in cells in intermediate states that have both, epithelial and mesenchymal characteristics [44]. This process occurs naturally during embryogenesis, and organ development [45] in adult life participates in diverse process like tissue regeneration and organ fibrosis but this reversible program reactivates during cancer pathogenesis [46-48].
During the EMT process, epithelial cells progressively lose their classic appearance by the disruption of cell-cell junctions, degradation of the underlying basement membrane, and reorganization of the extracellular matrix (ECM). After that, the cells adopt a mesenchymal morphology with the particularity of reverting back to an epithelial state using the reverse process, known as mesenchymal-epithelial transition (MET) [49]. Recently evidence showed that the reactivation of EMT in neoplastic cells is responsible for the malignant progression of possibly all types of cancer (pancreatic, hepatocellular, blander, lung, colorectal carcinoma) [50-55].
These morphology changes are possible by the interaction of tumoral cells and diverse types of cells like angiogenic vascular cells, tumors, cancer-associated fibroblasts (CAFs) and immune cells like tumor associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs), natural killer (NK) cells, and different T lymphocyte subpopulations, such as CD4+ T cells, CD8+ T cells, regulatory T cells (Tregs), and B lymphocytes mediated by signals pathways and transcription factors in the tumoral microenvironment (TME) [56,57].
The transcription factors (TFs) are involucrate in cellular functions such as cell proliferation, migration, apoptosis, and extracellular degradation, and their activation can occur by a variety of stimuli like extracellular signals, external agents, and pathologic intrinsic states [58-60]. TFs participate during the carcinogenesis process stimulate, tumor progression, angiogenesis, immunosuppression, invasion, and metastasis. In particular, has linked increased expression of EMT TFs such as TWIST1, SNAIL1, and ZEB1 with immune evasion during the EMT process. In the early stages of transformation, cytokines/ chemokines (TGFβ, IL-6, EGF, VEGF, and HGF) secreted by CAFs and TAMs attract various stromal and immune cells to the TME. These immune cells in turn provide a niche that facilitates the epithelial-mesenchymal transition, this occurs by the inhibition of effector immune cells and the accumulation of myeloid suppressive cells [61-64].. Another example of EMT TFs that recently have been associated with immune evasion is brachyury (T-box transcription factor T) which in a normal state is correlated with development pathways [65]. When is overexpressed by tumoral cells, upregulates levels of the transmembrane glycoprotein mucin-1 (MUC-1). Overexpression of MUC-1 led to reduced susceptibility to killing by tumor necrosis-related apoptosis-inducing ligand (TRAIL) and to CD8+ cytotoxic T cells (CTLs) lysis [66]. In addition, a study realized in diverse types of carcinomas demonstrates that high levels of brachyury reduce the susceptibility of carcinoma cells to either CTLs, NK, and lymphokine-activated killer (LAK) cells by decreasing the contribution to cell death of caspase-dependent pathways [67]. The phenotypic alterations that the tumoral cells suffer during the EMT process have significant consequences for the recognition by cells of the immune system and down or upregulation of cell surface molecules of immunological significance have been described [68]. In melanoma cells have been reported that EMTlike alterations reduce the expression of diverse tumor antigens allowing the escape from T cells specific for these antigens [69]. T cell-driven immunoediting of breast tumors in Neu-transgenic mice led to the emergence of antigen-loss variants that had undergone an EMT [70]. Furthermore, cells of non-small cell lung cancer (NSCLC) showed a significantly reduced expression of immunoproteasome components and their regulators [71]. The antigenic peptides are produced by the immunoproteasome and bind to human leukocyte antigen (HLA)-I molecules for recognition by CD8+ T cells. Consequently, the low expression of the immunoproteasome leads to decreased presentation of antigenic peptides. The reduction in antigen presentation and recognition by CTLs have been correlated with the downregulation of HLA-1 molecules demonstrated in epithelial cell lines of different tumors because of EMT [72,73]. The alteration in the levels of immune effectors like CTLs had clinical consequences, studies realized in mesenchymal subtype of ovarian cancer demonstrated worse prognosis and survival compared to other subtypes, this was correlated with a decreased number of CD8+ tumor-infiltrating lymphocytes (TILs) [74], other findings realized in an immunohistochemistry study performed in patients with gastric cancer, had a high expression of EMT traits, the infiltration of TAMs, and the expression of TGF-β1 were associated with a negative prognosis [75]. In a study of lung adenocarcinoma [76], EMT markers were associated with enhanced tumor infiltration of CD4+Foxp3+ Tregs and upregulation of inhibitory immune checkpoint molecules such as programmed cell death (PD)-ligand (L) 1, PD-L2, and CTLA-4, as well as a study in patients with adenocarcinoma of the lung, showed an association between EMT tumor cells and increased numbers of infiltrating PD-1+ cells [69]. Recent associations between autophagy and EMT resistance to immune effector mechanisms were made [69]. Studies realized in breast cancer cells showed that tumoral cells reduce the susceptibility to CTL-mediated lysis depending on upregulation of the stem cell marker Kruppel-like factor-4 (KLF-4) and miR-7 down regulation [77,78] also this type of tumoral cell that acquires an EMT phenotype had an increase resistance to CTL-mediated lysis, these resistant cells exhibited attenuation in the formation of an immunologic synapse with CTLs along with induction of autophagy [79]. Therefore, autophagy appeared to be critical to resistance to CTL-mediated lysis and a main process for EMT. Thereby, diverse studies had demonstrated that EMT is associated with the upregulation of inhibitory immune checkpoint molecules such as programmed cell death (PD)-ligand (L) 1, PD-L2. A study in patients with adenocarcinoma of the lung showed an association between EMT tumor cells and increased numbers of infiltrating PD-1+ cells [69]. Another study had demonstrated that CD4+ T cells are the main source of IL-6 in clear cell renal carcinoma cells can induce EMT- like features [80].
The role of proteins
The activity of diverse pathways and their transcription factors during the conservation of the tissues requires the function of a variety of proteins as shown in Table 1, because of the variety of activities of these proteins that are recently associated with the EMT process and the immune response, this is a recent area of research, and we will talk about this interaction:
E-cadherin: E-cadherin is a transmembrane glycoprotein and its main function is structural, maintaining cell–cell interactions and the polar orientation of cells [81]. Deletion and dysregulation of E-cadherin are correlated with alteration of intercellular junction, and subsequently, acquire a more mobile and invasive mesenchymal-like phenotype, this allows tumor invasion and metastasis [82,83]. In view of these facts, previously E-cadherin was considerate a cause of EMT [73], however, the abatement of E-cadherin is more a consequence of EMT than a cause [84,85].
In the normal tissues E-cadherin maintain intercellular union binding with β- and p120-catenins yielding a multiprotein complex, which interacts through α-catenin to actin filaments in the cell [86]. In normal cells, E-cadherin exerts its tumor-suppressing role mainly by sequestering β-catenin from its binding to LEF (lymphoid enhancer factor)/TCF (T cell factor) which has the function of transcribing genes of the proliferative Wnt signaling pathway [87].
Several studies have demonstrated that E-cadherin is involved in several signaling pathways, such as the Wnt/β-catenin, Rho GTPase, and EGF/EGFR pathways [71,72,88] which are activated in carcinogenesis and play a role in many cancers. Therefore, alteration of these pathways had been correlated with the partial or complete loss of E-cadherin.
Also, CDH1, gen which codes for E-cadherin, is susceptible to a wide range of mutations that are related to the decrease of this protein [89-91]. The alterations on the expression of Ecadherin has big impact on the EMT process, thereby the mutations in CDH1 are also important, the alterations of this gene are implicated in hereditary carcinomas like the diffuse gastric and lobular breast cancer [92,93]. CDH1 repress the transcription of E-cadherin by specific molecules that bind to specific DNA sequences of the E boxes of CDH1 promoter, activating secondarily mesenchymal genes that promotes the EMT process. This process of losing epithelial gene expression and activation of mesenchymal genes involves the transcription factors SNAIL, ZEB, and TWIST [94-97]. Studies realized in breast carcinoma reported that silence SNAIL increases the expression of E-cadherin and decrease the expression of mesenchymal markers, also high levels of ZEB1 were associated with high aggressive carcinoma and advanced disease stage [ 98,99].
E-cadherin is considered as an invasion suppressor, and its deregulation is often found in advanced cases of some epithelial carcinomas [100].. The tumor microenvironment is also implicated in the dysregulation of E-cadherin by the NF-κB pathway that mediates the activation of cytokines IL-1, IL-6, IL-8, MCP-1, apoptotic factors (Ciap, c-FLIP, BCL-XL), vascular endothelial factor (VEGF) and matrix metalloproteinases 2 and 9 (MMP-2 and MMP-9) in normal and malignant cells. The E-cadherin/catenin complex is known to decrease the activity of nuclear factor kappa B (NF-κB) [101,102]. The signaling pathway that involves NFκB regulates the epithelial cell phenotype during inflammation, which is associated with carcinogenesis [103-105].
Fibronectin-1: Fibronectin 1 (FN-1) was originally described like a structural protein that is present on the cell surfaces [106]. We know that this protein is implicated in more processes such as wound healing, embryogenesis and blood coagulation [107].
Like other proteins, the expression of FN-1 is upregulated in several cancers (melanoma, ovarian, hepatocellular [108-112] but interestingly the role of FN-1 is very variant. On the one hand is associated with a decrease level of E-cadherin and an increase in N-CAD, Vimentin, and other TFs associated with EMT and metastasis [113]. On the other side, FN-1 showed evidence of inhibiting tumor growth and metastasis. Glasner, A. et al. [114] showed that in an early pre-metastatic stage the tumor “prepares” itself for dissemination, this is detected by the NK cells via NCR1 mediated secretion of IFN-γ and FN-1 modifying tumor organization and consequently restricting metastasis [114].
The variability in FN-1 activity is also correlated with its ubication, evidence shows that endogenous FN-1 was associated with metastasis and poor prognosis in advanced stages [108- 110]in contrast when is expressed in form of deposits into ECMs in the immunosuppressive tumor microenvironments (TIEMs) where promotes early tumor progression but curiously in these cases, the patients had a better prognosis [111,112].
The colony-stimulating factor 1 (CSF-1) has been shown to be a regulator of endogenous cellular FN expression [115, 116], also the activation of HIFs (HIF-1, HIF2) [117-119] have been correlated with the endogenous synthesis of FN-1 and as well-known HIFs has an important role as a stimulator for the tumor cells to change epithelial to a mesenchymal phenotype [120]. Thereby FN-1 can be overexpressed in tumor cells under hypoxic conditions. Meanwhile, SPARC that which is a protein implicated in the dormancy process was correlated with the increased levels of periFN, therefore, the effect of SPARC on tumor dormancy may be due to the elevated level of periFN assembly [105].
The Programmed cell death protein 1 (PD-1) and their ligand, programmed cell death ligand 1 (PD-L1) which work as a co-inhibitory factor of the immune response, plays an important role in various malignancies where it can attenuate the host immune response to tumor cells [121]. miR200/ZEB1 axisinduced EMT pathways and regulates PD-L1 expression in CTS and FN expression is evidenced when miR200 is overexpressed or ZEB1 mRNA is stabilized [122-124], indicating the possibility that periFN on CTCs exerts PD-1/PD-L1-mediated immune checkpoint to escape T-cell-mediated cytotoxicity [125].
The expression of TFs like Prrx1, Snail 1 or Twist 1 enhances FN expression and promote mestastasis [125]. In a study realized in Ovarian cancer was found that TWIST-1 promoted metastasis discoid in domain receptor, increase the activity of matrix modeling enzymes, and the cleavage of FN-1, leading to elevated migratory and invasive activities of tumor cells [113]. Snail1, p65NF-kB, and PARP1 cooperate to activate the expression of FN-1 in cells undergoing EMT [126]. In a study realized in cells of thyroid cancer that shows increased levels of FN-1 expression in aggressive forms of this tumor, also have increased levels of Snail and p65NF-Kb [127].
SPARC: SPARC (secreted protein acidic and cysteine rich) is a multifunctional calcium-binding glycoprotein, the principal function of this matricellular molecule is regulate the interactions between cells and their surrounding extracellular matrix, is implicated in cell functions such as proliferation, migration and cell shape, SPARC also influence pathways associated with development, wound healing, tissue remodeling, angiogenesis, apoptosis, and regulation of the immune response [128-131]. SPARC could be expressed on the cell surface and within the intracellular compartment, when is expressed extracellular SPARC functions as a matricellular protein, while is intracellular and membrane‐associated SPARC regulate cellular apoptotic pathways [132].
Dysregulation of SPARC has been found in the tumorogenesis process, thereby has correlated with the cell cycle progression, angiogenesis, apoptosis, EMT process, and metastasis [133- 136]. SPARC works as a double agent in cancer; thus, increased or decreased levels of this protein have been found in diverse tumor. Higher levels of SPARC have been reported in breast cancer, hepatocellular and melanoma [137-139], however, lower levels of SPARC have been found in ovarian, prostatic, colorectal, and pancreatic cancers [140-142]. The function of SPARC is highly heterogeneous among tumor types, because SPARC has tissue specificity, and even in the same carcinoma as pancreatic cancer could act as a tumor suppressor or an oncogene [143- 146], the overexpression of SPARC could act as a tumor suppressor gene, inhibiting proliferation, invasion and angiogenesis in cancers such as gastric and ovarian cancer [147]. On the other hand, low levels or absent expression of SPARC is correlated with bad prognosis and aggressive clinicopathological features for example in endometrial and colorectal carcinoma [148,149]. It has been demonstrated that low or absent expression of this protein is a consequence of the methylation of the gene promoter region [150].
The paradoxical role of SPARC as a tumor suppressor or promoter may be due to the specific microenvironment of each type of cancer [135]. The tumor associated stroma mainly express SPARC, predominantly in the cytoplasm and ECM of stromal fibroblasts. The high expression of SPARC on CAF supports the idea that stromal SPARC enhances tumor growth and tumor–stroma interactions, contributing to a more aggressive malignancy [151].
SPARC is also correlated with the TGF‐β/Smad 2/3 pathway which regulates the synthesis of diverse ECM proteins. SPARC is a downstream mediator of the transcription factor SNAIL which is a regulator of TGF‐β1 [152]. By the way TGF‐β1 is implicated in the formation and dysregulation of EMT. SPARC stimulates the expression of TGF-B1 in tumor cells such as the pancreatic cancer cells [153]. The SPARC-TGFβ interaction affects negatively the expression levels of the epithelial markers such as: E-cadherin, Integrin β1 and Focal Adhesion Kinase (FAK) and increases the expression of mesenchymal markers N-cadherin and vimentin, which facilitates the EMT and metastasis processes [154].
HLA-E: Major histocompatibility complex (MHC) class I molecules have main functions in both the innate and the adaptive immune system through the presentation of peptides from intracellular proteins to lymphocytes and by acting as ligands for NK cell receptors, this molecules can be classified in two groups, the non-classical HLA class Ib family (HLA-E,F,G and H) which are homologous to classical HLA Ia molecules (HLA-A,B,C) but their difference is the limited polymorphism, low cell expression, and limited tissue distribution [155].
The Human leukocyte antigen-E (HLA-E) belongs to a nonclassical HLA class Ib group of molecules. In contrast to the other nonclassical class I molecules which are mainly restricted to specific tissues, e.g., placenta [156], expression of HLA-E is more ubiquitous and virtually every healthy cell in the body positive for HLA class I also expresses HLA-E but their expression levels are relatively low compared with class Ia. Other mains differences of HLA-E is the least polymorphic of all the MHC class I molecules and can interact with inhibitory and activating receptors present in NK cells and T cells, having a dual function in the immune system [157,158].
The dual activity of HLA-E makes that the effect of this molecule and its alterations on the cellular immune response is too complex. The immune activation or suppression depends of the receptor and the responding cell, one of the most important receptor family interacting with HLA-E is the family of CD94/ NKG2A which is expressed on NK cells. Two members of this family are NKG2A and NKG2C which forms a heterodimer with CD94. The interaction HLA-E and CD94/NKG2A provide an inhibitory signal to the NK cells meanwhile the binding of HLA-E and CD94/NKG2C delivers an activation signal to the NK cells. HLA-E has a major affinity for NGK2A which means a priority inhibitory function [159-161].
Usually, in cancer, HLA class Ia molecules are lost, this allows tumor cells to prevent T cell mediated recognition [162]. Studies had found that many tumors could maintain expression of HLA-E even in the absence of classical HLA class Ia molecules, suggesting a predominantly immunosuppressive role for HLA-E in anti-tumor immunity [163].
Some investigations had demonstrated high levels of HLA-E in various cancer types as gynecological cancers, renal, stomach and colorectal cancer [164-166] suggesting that this altered level functions as an acquired resistance mechanism after immune activation in the tumor microenvironment [167].
Recent findings associate HLA-E with a worse prognosis, for relapse-free survival in cancers like ovarian and breast carcinoma but interestingly only when the classical expression was lost [168,169]. On the other hand, retained expression of classical HLA class Ia was associated with a better prognosis for patients with cervical carcinoma and non–small cell lung carcinoma (NSCLC). However, this predictive value is abrogated when tumors display high HLA-E levels, suggesting that HLA-E mediates resistance against CD8 T-cell attack [170-172]. Therefore, HLA-E expression seems to be dependent on immune microenvironment.
C- terminal tensin-like (TNS4): C-terminal tensin-like (TNS4) is also known as COOH-terminus tensin-like molecule (CTEN) belong to the tensin gene family, which is formed by other 3 members (TNS1, TNS2 and TNS3) [173]. Tensins play important roles in various biological processes, such as cell adhesion, proliferation, migration invasion and apoptosis [174-176].
TNS4 promotes cell migration by triggering the uncoupling of integrins from the actin cytoskeleton [177], interestingly the expression of this protein is up-regulated or downregulated in different types of cancers, suggesting that alterations on TNS4 plays a critical role in tumorigenesis, even more important TNS4 could have both, oncogenic and tumor suppressor functions [178-180].
Up regulation of TNS4 was found in lung, colon, stomach, pancreatic and breast cancer and was also correlated with poor prognosis [181,182] on the other hand, downregulation of TNS4 was detected in the prostate (where normally is high expressed [183] and kidney cancers [184-186]. The specific mechanisms of this process of dysregulation are poorly described.
Evidence demonstrates that TNS4 bind with c-Cbl, a ubiquitin ligase that promotes the degradation of active β-catenin [187]. This protein forms part of the Wnt/β-catenin signaling which is implicated in facilitates cancer stem cell renewal, cell proliferation and differentiation [188], β-Catenin indeed regulated the expression of VEGFA in colon cancer, VEGFA is an essential growth factor for vascular endothelial cells in normal and pathophysiological conditions [189]. Another study demonstrated protein expression levels of VEGFA were found to be upregulated in different subtypes of BRCA, inversely correlating with CTEN expression [190].
This activity may be related with the role of TNS4 as oncogene. Other studies had found that in invasive breast cancer TNS4 was associated with EGFR and HER2, also enhances the transcriptional activity of STAT3, contributed to the invasion and metastasis process of breast cancer cells [191]. Another study revealed that TNS reduces the E-cadherin levels enhanced EMT process [192].
Furthermore, investigations in non-small cell lung cancer shows that levels of TGF-β1 are both significantly correlated with NSCLC tumor size, this study found that overexpression of CTEN promotes TGF-β1 expression level and that overexpression of TNS4 promoted migration and invasion of human lung adenocarcinoma secondarily to TNS4 upregulated N-cadherin and Vimentin level while downregulated E-cadherin level [193].
In gastric cancer cells studies had found that tensin 4 can interact with PI3K through the SH2 domain to activate PI3K/ AKT signaling pathways which produce p-GSK3β, this increases the expression level of Snail by promoting its nuclear translocation [194,195]. Thus, TNS4 was closely associated with tumor infiltration depth, lymph node metastasis, and distal metastasis [196]. Tensin 4 may regulate EMT to promote invasion and migration of GC cells through the AKT/GSK-3β/Snail signaling pathway, providing a potential target for GC treatment [197] (Figure 1B).
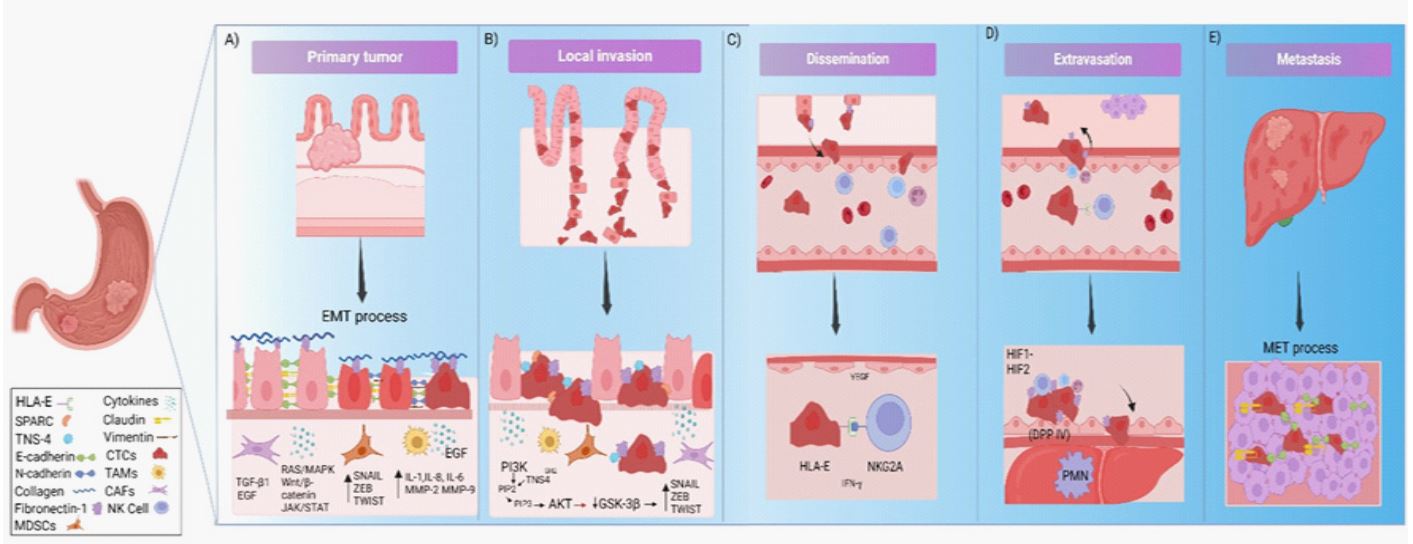
Table 1: The principal function of E-Cadherin, Fibronectin-1, Tensin-4, SPARC, HLA-E, and their principal sites of expression in normal conditions.
| Protein | Function | Tissues with higger expression | Tissues with lower expression |
|---|---|---|---|
| E- cadherin |
Cell-cell adhesión. Maintenance of normal tissue architecture. |
Detected in many tissues. Enhanced in epithelial tissues. |
Neuronal tissue. |
| Fibronectin-1 |
Cell adhesion, migration, growth, differentiation. Maintenance of cell shape. Wound healing. |
Detected in many tissues. Enhanced liver and placenta. |
Brain. |
| Tensin-4 |
Cell migration. Cartilage development. Glandular duct morphogenesis. Apoptosis. Linking signal transduction pathways to the cytoskeleton. |
Detected in some tissues like skin and gastrointestinal tract. |
Respiratory system. Connective and soft tissue. |
| SPARC |
Regulate the interactions between cells and their
surround- ing extracellular matrix. Cell proliferation, migration and cell shape. Wound healing. Tissue remodeling. Regulation of the immune response. |
Detected in all tissues, enhanced in pla- centa, brain and kidney. |
Muscular tissues. Gastrointestinal tract. |
| HLA-E |
Presentation of peptides from intracellular proteins to
lym- phocytes. Ligands for NK cell receptors. |
Detected in many tissues, enhanced in lung, kidney, lymph nodes. |
Muscular tissues (Heart muscle, Smooth muscle, Skeletal muscle) Adipose tissue. |
Conclusion
We have discussed the fundamentals of immune evasion and the crosstalk between EMT tumor cells and immune cells, while this concept is quite clear, there remain important unresolved questions about their interaction. First, as already discussed, tumoral cells use diverse mechanisms to elucidate the immune response jointly known as immunoediting. Second, we presented the relationship between the EMT process and immune response, where the cyclical relationship is mediated by TFs such as TWIST, SNAIL1, and new ones like brachyury, the final products of these TFs are a reduced susceptibility to killing by CD8+- cytotoxic T cells lysis, reduction in antigenic presentation and recognition by CTLs, upregularion of inhibitory immune checkpoint molecules such as programmed cell death (PD)-ligand and stimulation of other mechanisms such as autophagy. Third, we do not know all the molecules that participate in this interaction, we present in this review the ones that are important in each stage of the EMT and immune evasion. We have seen that some of these proteins like FN-1 have different clinical result apparently on a level-dependent way, even in the same tissue, this could be mediated by the interaction between EMT, TME and immune response but this field have not been investigated so far. Much work remains to be done in this field, combined consideration of these processes might stimulate new studies and result in novel discoveries in the future.
Declarations
Funding: This review did not recive any specific grant from funding agencies in the public or commercial sectors.
Disclosures: The authors declare that there is no conflict of interest.
References
- International Agency for Research on Cancer. Global Africa Latin America and the Caribbean Northern America Asia Europe Oceania Data source: GLOBOCAN 2020 Graph production: Global Cancer Observatory, (hyperlinked with http://gco.iarc.fr/ )
- Eccles SA, Welch DR.Metastasis: recent discoveries and novel treatment strategies. Lancet 2007; May 19;369(9574):1742-57. doi: 10.1016/S0140-6736(07)60781-8.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. CellMar 2011; 4;144(5):646-74. doi: 10.1016/j.cell.2011.02.013.
- Fouad YA, Aanei C. Revisiting the hallmarks of cancer. Am J Cancer Res. 2017; May 1;7(5):1016-1036.
- Abbott, M., & Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Seminars in Oncology Nursing, 2019; 35 (5) 150923 doi: 10.1016/j.soncn.2019.08.002.
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med 2010; 363(8):711–23
- Thompson RH, Gillett MD, Cheville JC, Lohse CM, Dong H, et al. Costimulatory B7-H1 in renal cell carcinoma patients: indicator of tumor aggressiveness and potential therapeutic target. PNAS 2004; 101(49):17174–79
- Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, et al. (). Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. PNAS 2007; 104(9):3360–65
- Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csoszi T, et al. Pembrolizumab versus ˝ chemotherapy for PD-L1-positive nonsmall-cell lung cancer. N. Engl. J. Med. 2016; 375(19):1823–33
- Zhang Y, Zheng J. Functions of Immune Checkpoint Molecules Beyond Immune Evasion. Adv Exp Med Biol 2020; 1248:201-226.
- Topalian, F.S. Hodi, J.R. Brahmer, S.N. Gettinger, D.C. Smith, D.F. McDermott, et. al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer, N Engl J Med 2012; 366 2443-2454.
- Dongre A, Rashidian M, Reinhardt F, Bagnato A, Keckesova Z, Ploegh HL and Weinberg RA. Epithelial-to-mesenchymal transition contributes to immunosuppression in breast carcinomas. Cancer Res, Advance online publication 2017; 77,15: 3982-3989.
- Herzberg, B., Campo, M. J., & Gainor, J. F. Immune Checkpoint Inhibitors in Non-Small Cell Lung Cancer. The oncologist, 2017; 22(1), 81–88. https://doi.org/10.1634/theoncologist.2016-0189
- Xu, J. W., Wang, L., Cheng, Y. G., Zhang, G. Y., Hu, S. Y., Zhou, B., & Zhan, H. X. Immunotherapy for pancreatic cancer: A long and hopeful journey. Cancer letters 2018; 425, 143–151. https://doi.org/10.1016/j.canlet.2018.03.040
- Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J and Harlin H. Immune resistance orchestrated by the tumor microenvironment. Immunol Rev 2006; 213(1), 131–145.
- Mittal V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol 2018 Jan 24; 13:395-412. doi: 10.1146/annurev-pathol-020117-043854.
- Jiang, Y., & Zhan, H. Communication between EMT and PD-L1 signaling: new insights into tumor immune evasion. Cancer Letters 2019; 468 (1), 72-81. doi: 10.1016/j.canlet.2019.10.013
- Tran TB, Maker VK, Maker AV. Impact of Immunotherapy after Resection of Pancreatic Cancer. J Am Coll Surg. 2019 Jul;229(1):19-27.e1. doi: 10.1016/j.jamcollsurg.2019.01.016.
- Bray-French K, Hartman K, Steiner G, Marban-Doran C, Bessa J, Campbell N, et al. Managing the Impact of Immunogenicity in an Era of Immunotherapy: From Bench to Bedside. J Pharm Sci. 2021; Jul;110(7):2575-2584. doi: 10.1016/j.xphs.2021.03.027.
- Franciosi, J. R., Gelmini, G. F., Roxo, V. S., de Carvalho, N. S., & Bicalho, M. D. G. Is there a role played by HLA-E, if any, in HPV immune evasion? Scandinavian journal of immunology, 2020; 91(3), e12850. doi: 10.1111/sji.12850
- Qadir, F., Aziz, M. A., Sari, C. P., Ma, H., Dai, H., Wang, X., Raithatha, D, et al. Transcriptome reprogramming by cancer exosomes: identification of novel molecular targets in matrix and immune modulation. Molecular cancer, 2018; 17(1), 97. https://doi.org/10.1186/s12943-018-0846-5
- Bure, I. V., Nemtsova, M. V., & Zaletaev, D. V. Roles of E-cadherin and Noncoding RNAs in the Epithelial-mesenchymal Transition and Progression in Gastric Cancer. International journal of molecular sciences, 2019; 20(12), 2870. doi: 10.3390/ijms20122870
- Corthay A. Does the immune system naturally protect against cancer? Frontiers in immunology, 2014; 5(1), 197. doi:10.3389/fimmu.2014.00197
- Burnet M. Cancer: a biological approach. III. Viruses associated with neoplastic conditions. IV. Practical applications. British medical journal 1957;1(5023), 841–847. Doi: 10.1136/bmj.1.5023.841
- Dunn, Gavin P.; Bruce, Allen T.; Ikeda, Hiroaki; Old, Lloyd J.; Schreiber, Robert D. Cancer immunoediting: from immunosurveillance to tumor escape, Nat Immunology 2002; 3(11), 991–998. doi:10.1038/ni1102-991
- Dunn, G. P., Old, L. J., & Schreiber, R. D. The three Es of cancer immunoediting. Annual review of immunology, 2004; 22, 329–360. https://doi.org/10.1146/annurev.immunol.22.012703.104803
- Schreiber, R. D., Old, L. J. & Smyth, M. J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011; 331(1), 1565–1570.
- Mohme, M., Riethdorf, S., & Pantel, K. Circulating and disseminated tumour cells — mechanisms of immune surveillance and escape. Nature Reviews Clinical Oncology, 2016; 14(3), 155- 167. doi:10.1038/nrclinonc.2016.144.
- Jacobs, J. F., Nierkens, S., Figdor, C. G., de Vries, I. J., & Adema, G. J. Regulatory T cells in melanoma: the final hurdle towards effective immunotherapy? The Lancet. Oncology, 2012; 13(1), e32–e42. https://doi.org/10.1016/S1470-2045(11)70155-3
- Yokokawa, J., Cereda, V., Remondo, C., Gulley, J. L., Arlen, P. M., Schlom, J., & Tsang, K. Y. Enhanced functionality of CD4+CD25(high)FoxP3+ regulatory T cells in the peripheral blood of patients with prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research, 2008; 14(4), 1032–1040. https://doi.org/10.1158/1078-0432.CCR-07-2056
- Gasparoto, T. H., de Souza Malaspina, T. S., Benevides, L., de Melo, E. J., et al. Patients with oral squamous cell carcinoma are characterized by increased frequency of suppressive regulatory T cells in the blood and tumor microenvironment. Cancer immunology, immunotherapy 2010; 59(6), 819–828. doi: 10.1007/s00262-009-0803-7
- Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nature reviews. Immunology, 2006;6(4), 295–307. doi: 10.1038/nri1806
- Vinay, D. S., Ryan, E. P., Pawelec, G., Talib, W. H., Stagg, J., Elkord, E., Kwon, B. S. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Seminars in Cancer Biology, 2015;35(1), 185- 198. doi: 10.1016/j.semcancer.2015.03.004.
- Maeurer, M. J., Gollin, S. M., Martin, D., Swaney, W., Bryant, J., Castelli, C., et al. Tumor escape from immune recognition: lethal recurrent melanoma in a patient associated with downregulation of the peptide transporter protein TAP-1 and loss of expression of the immunodominant MART-1/Melan-A antigen. The Journal of clinical investigation, 1996; 98(7), 1633–1641. Doi: 10.1172/JCI118958
- Lind, M. H., Rozell, B., Wallin, R. P., van Hogerlinden, M., Ljunggren, H. G., Toftgård, R., & Sur, I. Tumor necrosis factor receptor 1-mediated signaling is required for skin cancer development induced by NF-kappaB inhibition. Proceedings of the National Academy of Sciences of the United States of America, 2004; 101(14), 4972–4977. https://doi.org/10.1073/pnas.0307106101
- Lin, E. Y., Gouon-Evans, V., Nguyen, A. V., & Pollard, J. W. The macrophage growth factor CSF-1 in mammary gland development and tumor progression. Journal of mammary gland biology and neoplasia, 2002; 7(2), 147–162. https://doi.org/10.1023/a:1020399802795.
- Klein, S. C., Jücker, M., Abts, H., & Tesch, H. IL6 and IL6 receptor expression in Burkitt’s lymphoma and lymphoblastoid cell lines: promotion of IL6 receptor expression by EBV. Hematological oncology, 1995;13(3), 121–130. doi: 10.1002/hon.2900130302.
- Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med 1996; 2:1096–103.
- Raza, A., Franklin, M. J., & Dudek, A. Z. Pericytes and vessel maturation during tumor angiogenesis and metastasis. American Journal of Hematology, 2010; 85(8), 593–598. doi:10.1002/ajh.21745
- Cooke VG, LeBleu VS, Keskin D, Khan Z, O’Connell JT, Teng Y, et al. Pericyte depletion results in hypoxia-associated epithelialto-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012; 21:66–81.
- Maeda H, Shiraishi A. TGF-beta contributes to the shift toward Th2-type responses through direct and IL-10-mediated pathways in tumor-bearing mice. J Immunol; 1996; 156:73–8.
- Driessens, G., Kline, J., & Gajewski, T. F. Costimulatory and coinhibitory receptors in anti-tumor immunity. Immunological reviews, 2009; 229(1), 126–144. Doi:10.1111/j.1600-065X.2009. 00771.x
- Topalian SL, Drake CG, Pardoll DM. Targeting PD-1/B7-H1 (PDL1) pathway to activate antitumor immunity. Curr Opin Immunol; 2012; 24:207–12.
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009 Jun;119(6):1420-8. doi: 10.1172/JCI39104.
- Hay, E.D. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev. Dyn. 2005; 233:706–720.
- Strutz, F., et al. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation. Kidney Int. 2002; 61:1714–1728
- Zeisberg, M., Yang, C., Martino, M., Duncan, M. B., Rieder, F., Tanjore, H., & Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. The Journal of biological chemistry, 2007; 282(32), 23337–23347. Doi:10.1074/jbc.M700194200
- Zeisberg, M., Hanai, J., Sugimoto, H., Mammoto, T., Charytan, D., Strutz, F., & Kalluri, R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nature medicine, 2003; 9(7), 964–968. Doi:10.1038/nm888
- Baumgart, E., Cohen, M. S., Silva Neto, B., Jacobs, M. A., Wotkowicz, C., Rieger-Christ, K. M., Biolo, A., Zeheb, R., Loda, M., Libertino, J. A., & Summerhayes, I. C. Identification and prognostic significance of an epithelial-mesenchymal transition expression profile in human bladder tumors. Clinical cancer research: an official journal of the American Association for Cancer Research 2007; 13(6), 1685–1694. Doi: 10.1158/1078-0432.CCR-06-233
- Gravdal, K., Halvorsen, O. J., Haukaas, S. A., & Akslen, L. A. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clinical cancer research: an official journal of the American Association for Cancer Research, 2007; 13(23), 7003–7011. https://doi.org/10.1158/1078-0432.CCR-07-1263
- Rhim, A. D., Mirek, E. T., Aiello, N. M., Maitra, A., Bailey, J. M., McAllister, F., et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012; 148(1-2), 349–361. Doi: 10.1016/j.cell.2011.11.025
- Lee, T. K., Poon, R. T., Yuen, A. P., Ling, M. T., Kwok, W. K., Wang, X. H., et al. Twist overexpression correlates with hepatocellular carcinoma metastasis through induction of epithelial-mesenchymal transition. Clinical cancer research 2006; 12(18), 5369–5376. doi.10.1158/1078-0432.CCR-05-2722
- Mahmood, M. Q., Ward, C., Muller, H. K., Sohal, S. S., & Walters, E. H. (). Epithelial mesenchymal transition (EMT) and non-small cell lung cancer (NSCLC): a mutual association with airway disease. Medical oncology 2017; 34(3), 45. Doi: 10.1007/s12032-017-0900-y
- Prudkin, L., Liu, D. D., Ozburn, N. C., Sun, M., Behrens, C., Tang, X., Brown, K. C., Bekele, B. N., Moran, C., & Wistuba, I. Epithelial-to-mesenchymal transition in the development and progression of adenocarcinoma and squamous cell carcinoma of the lung. Modern pathology 2009; 22(5), 668–678. https://doi.org/10.1038/modpathol.2009.19
- Shioiri, M., Shida, T., Koda, K., Oda, K., Seike, K., Nishimura, M., Takano, S., & Miyazaki, M. Slug expression is an independent prognostic parameter for poor survival in colorectal carcinoma patients. British journal of cancer 2006; 94(12), 1816–1822. Doi: 10.1038/sj.bjc.6603193
- Shintani, Y., Fujiwara, A., Kimura, T., Kawamura, T., Funaki, S., Minami, M., & Okumura, M. IL-6 Secreted from Cancer-Associated Fibroblasts Mediates Chemoresistance in NSCLC by Increasing Epithelial-Mesenchymal Transition Signaling. Journal of thoracic oncology 2016; 11(9), 1482–1492. doi: 10.1016/j.jtho.2016.05.025
- Yu, Y., Xiao, C. H., Tan, L. D., Wang, Q. S., Li, X. Q., & Feng, Y. M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-β signalling. British journal of cancer, 2014; 110(3), 724–732. Doi:10.1038/bjc.2013.768
- Brabletz, T., Kalluri, R., Nieto, M. A., & Weinberg, R. A. (2018). EMT in cancer. Nature reviews. Cancer, 18(2), 128–134. doi: 10.1038/nrc.2017.118
- Kahlert, U. D., Joseph, J. V., & Kruyt, F. A. E. EMT- and MET-related processes in nonepithelial tumors: importance for disease progression, prognosis, and therapeutic opportunities. Molecular oncology 2017; 11(7), 860–877. https://doi.org/10.1002/1878-0261.12085
- Nieto, M. A., & Cano, A. The epithelial-mesenchymal transition under control: global programs to regulate epithelial plasticity. Seminars in cancer biology 2012; 22(5-6), 361–368. Doi: /10.1016/j.semcancer.2012.05.003
- Singh, S., & Chakrabarti, R. Consequences of EMT-Driven Changes in the Immune Microenvironment of Breast Cancer and Therapeutic Response of Cancer Cells. Journal of Clinical Medicine 2019;8(5), 642. doi: 10.3390/jcm8050642
- Brenot, A., Knolhoff, B. L., DeNardo, D. G., & Longmore, G. D. SNAIL1 action in tumor cells influences macrophage polarization and metastasis in breast cancer through altered GM-CSF secretion. Oncogenesis, 2018; 7(3). doi:10.1038/s41389-018-0042-x
- Cortés, M., Sanchez-Moral, L., de Barrios, O., Fernández-Aceñero, M. J., Martínez-Campanario, et al. Tumor-associated macrophages (TAMs) depend on ZEB1 for their cancer-promoting roles. The EMBO journal, 2017; 36(22), 3336–3355. Doi: 10.15252/embj.201797345
- Becht, E., Giraldo, N. A., Germain, C., de Reyniès, A., LaurentPuig, P., Zucman-Rossi, J., Dieu-Nosjean, M. C., Sautès-Fridman, C., & Fridman, W. H. Immune Contexture, Immunoscore, and Malignant Cell Molecular Subgroups for Prognostic and Theranostic Classifications of Cancers. Advances in immunology, 2016; 130, 95–190.DOI: 10.1016/bs.ai.2015.12.002
- Chen M, Wu Y, Zhang H, Li S, Zhou J and Shen J. The Roles of Embryonic Transcription Factor BRACHYURY in Tumorigenesis and Progression. Front. Oncol. 2020; 10:961. doi: 10.3389/fonc.2020.00961
- David, J. M., Hamilton, D. H., & Palena, C. MUC1 upregulation promotes immune resistance in tumor cells undergoing brachyury-mediated epithelial-mesenchymal transition. Oncoimmunology 2016; 5(4), e1117738. https://doi.org/10.1080/2162402X.2015.1117738.
- Hamilton, D. H., Huang, B., Fernando, R. I., Tsang, K. Y., & Palena, C. WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial-mesenchymal transition. Cancer research, 2014; 74(9), 2510–2519. https://doi.org/10.1158/0008-5472.CAN-13-1894
- Romeo, E., Caserta, C. A., Rumio, C., & Marcucci, F. The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System. Cells 2019; 8(5), 460. doi: 10.3390/cells8050460.
- Landsberg, J., Kohlmeyer, J., Renn, M., Bald, T., Rogava, M., Cron, M., Fatho, M., Lennerz, V., Wölfel, T., Hölzel, M., & Tüting, T. Melanomas resist T-cell therapy through inflammationinduced reversible dedifferentiation. Nature, 2012; 490(7420), 412–416. Doi: 10.1038/nature11538
- Woods, K., Pasam, A., Jayachandran, A., Andrews, M. C., & Cebon, J. Effects of epithelial to mesenchymal transition on T cell targeting of melanoma cells. Frontiers in oncology, 2014; 4, 367. doi: 10.3389/fonc.2014.00367
- Tripathi, S. C., Peters, H. L., Taguchi, A., Katayama, H., Wang, H., Momin, A., et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proceedings of the National Academy of Sciences of the United States of America, 2016; 113(11), 1555–1564. doi: 10.1073/pnas.1521812113
- López-Soto, A., Huergo-Zapico, L., Galván, J. A., Rodrigo, L., de Herreros, A. G., Astudillo, A., & Gonzalez, S. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. Journal of immunology 2013; 190(8), 4408–4419. doi: 10.4049/jimmunol.1202950.
- Chen, X.H.; Liu, Z.C.; Zhang, G.; Wei, W.; Wang, X.X.; Wang, H.; Ke, H.P.; Zhang, F.; Wang, H.S.; Cai, S.H.; et al. TGF-β and EGF induced HLA-I downregulation is associated with epithelialmesenchymal transition (EMT) through upregulation of snail in prostate cancer cells. Mol. Immunol 2015; 65, 34–42.
- Murakami, R., Matsumura, N., Mandai, M., Yoshihara, K., Tanabe, H., Nakai, H., et al. Establishment of a Novel Histopathological Classification of High-Grade Serous Ovarian Carcinoma Correlated with Prognostically Distinct Gene Expression Subtypes. The American journal of pathology 2016; 186(5), 1103–1113. Doi: 10.1016/j.ajpath.2015.12.029
- Yan, Y., Zhang, J., Li, J. H., Liu, X., Wang, J. Z., Qu, H. Y., Wang, J. S., & Duan, X. Y. High tumor-associated macrophages infiltration is associated with poor prognosis and may contribute to the phenomenon of epithelial-mesenchymal transition in gastric cancer. OncoTargets and therapy 2016; 9, 3975–3983. doi:10.2147/OTT.S103112
- Lou, Y., Diao, L., Cuentas, E. R., Denning, W. L., Chen, L., Fan, Y. H., Byers, L. A., Wang, J., Papadimitrakopoulou, V. A, et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clinical cancer research 2016; 22(14), 3630–3642. doi: 10.1158/1078-0432.CCR-15-1434
- Akalay, I., Janji, B., Hasmim, M., Noman, M. Z., André, F., De Cremoux, P., Bertheau, P., Badoual, C., Vielh, P., Larsen, A. K., Sabbah, M, et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer research, 2013; 73(8), 2418–2427. https://doi.org/10.1158/0008-5472.CAN-12-2432
- Akalay, I., Tan, T., Kumar, P. et al. Targeting WNT1-inducible signaling pathway protein 2 alters human breast cancer cell susceptibility to specific lysis through regulation of KLF-4 and miR-7 expression. Oncogene 2015; 34, 2261–2271 https://doi.org/10.1038/onc.2014.151
- Marcucci, F., & Rumio, C. How Tumor Cells Choose Between Epithelial-Mesenchymal Transition and Autophagy to Resist StressTherapeutic Implications. Frontiers in pharmacology 2018; 9, 714. https://doi.org/10.3389/fphar.2018.00714.
- Chen, Q. et al. Growth-induced stress enhances epithelial–mesenchymal transition induced by IL-6 in clear cell renal cell carcinoma via the Akt/GSK-3beta/ beta-catenin signaling pathway. Oncogenesis 2017; 6(8) e375. doi:10.1038/oncsis.2017.74
- Christofori, G., & Semb, H. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. Trends in biochemical sciences 1999; 24(2), 73–76. https://doi.org/10.1016/s0968-0004(98)01343-76
- Onder, T. T., Gupta, P. B., Mani, S. A., Yang, J., Lander, E. S., & Weinberg, R. A. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer research 2008; 68(10), 3645–3654. https://doi.org/10.1158/0008-5472.CAN-07-2938
- Kalluri, R., & Weinberg, R. A. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation 2009; 119(6), 1420–1428. https://doi.org/10.1172/JCI39104
- Chen, A., Beetham, H., Black, M. A., Priya, R., Telford, B. J., Guest, J., Wiggins, G. A., Godwin, T. D., Yap, A. S., & Guilford, P. J. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC cancer 2014; 14, 552. https://doi.org/10.1186/1471-2407-14-552
- Bure, I. V., Nemtsova, M. V., & Zaletaev, D. V. Roles of E-cadherin and Noncoding RNAs in the Epithelial-mesenchymal Transition and Progression in Gastric Cancer. International journal of molecular sciences, 2019; 20(12), 2870. https://doi.org/10.3390/ijms20122870.
- Ratheesh, A., & Yap, A. S. A bigger picture: classical cadherins and the dynamic actin cytoskeleton. Nature reviews. Molecular cell biology 2012; 13(10), 673–679. https://doi.org/10.1038/nrm3431
- Wong, S. H. M., Fang, C. M., Chuah, L. H., Leong, C. O., & Ngai, S. C. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Critical reviews in oncology/hematology 2018; 121, 11–22. https://doi.org/10.1016/j.critrevonc.2017.11.010
- Wang, Y., Shi, J., & Gong, L. Gamma linolenic acid suppresses hypoxia-induced gastric cancer cell growth and epithelial-mesenchymal transition by inhibiting the Wnt/b-catenin signaling pathway. Folia histochemica et cytobiologica 2020; 58(2), 117–126. https://doi.org/10.5603/FHC.a2020.0012
- Machado, J. C., Oliveira, C., Carvalho, R., Soares, P., Berx, G., Caldas, C., Seruca, R., Carneiro, F., & Sobrinho-Simöes, M. Ecadherin gene (CDH1) promoter methylation as the second hit in sporadic diffuse gastric carcinoma. Oncogene 2001; 20(12), 1525–1528. https://doi.org/10.1038/sj.onc.1204234
- Shargh, S. A., Sakizli, M., Khalaj, V., Movafagh, A., Yazdi, H., Hagigatjou, E., Sayad, A., Mansouri, N., Mortazavi-Tabatabaei, S. A., & Khorram Khorshid, H. R. (). Downregulation of E-cadherin expression in breast cancer by promoter hypermethylation and its relation with progression and prognosis of tumor. Medical oncology 2014; 31(11), 250. https://doi.org/10.1007/s12032-014-0250-y
- Liu, J., Sun, X., Qin, S., Wang, H., DU, N., Li, Y., Pang, Y., Wang, C., Xu, C., & Ren, H. CDH1 promoter methylation correlates with decreased gene expression and poor prognosis in patients with breast cancer. Oncology letters 2016; 11(4), 2635–2643. https://doi.org/10.3892/ol.2016.4274.
- Caldas, C., Carneiro, F., Lynch, H. T., Yokota, J., Wiesner, G. L., Powell, S. M., Lewis, F. R., Huntsman, D. G., Pharoah, P. D., Jankowski, J. A., et al. Familial gastric cancer: overview and guidelines for management. Journal of medical genetics, 1999; 36(12), 873–880.
- Corso, G., Figueiredo, J., La Vecchia, C., Veronesi, P., Pravettoni, G., Macis, D., Karam, R., Lo Gullo, R., et al. Hereditary lobular breast cancer with an emphasis on E-cadherin genetic defect. Journal of medical genetics, 2018; 55(7), 431–441. https://doi.org/10.1136/jmedgenet-2018-105337
- Peinado, H., Olmeda, D., & Cano, A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature reviews. Cancer, 2007; 7(6), 415–428. https://doi.org/10.1038/nrc2131
- Olmeda, D., Moreno-Bueno, G., Flores, J. M., Fabra, A., Portillo, F., & Cano, A. SNAI1 is required for tumor growth and lymph node metastasis of human breast carcinoma MDA-MB-231 cells. Cancer research, 2007; 67(24), 11721–11731. https://doi.org/10.1158/0008-5472.CAN-07-2318.
- Villarejo, A., Cortés-Cabrera, A., Molina-Ortíz, P., Portillo, F., & Cano, A. Differential role of Snail1 and Snail2 zinc fingers in Ecadherin repression and epithelial to mesenchymal transition. The Journal of biological chemistry, 2014; 289(2), 930–941. https://doi.org/10.1074/jbc.M113.528026
- Corso, G., Figueiredo, J., De Angelis, S. P., Corso, F., Girardi, A., Pereira, J., Seruca, R.,et al. E-cadherin deregulation in breast cancer. Journal of cellular and molecular medicine 2020; 24(11), 5930–5936. https://doi.org/10.1111/jcmm.15140
- Taki, M., Abiko, K., Ukita, M., Murakami, R., Yamanoi, K., Yamaguchi, K., Hamanishi, J., Baba, T., Matsumura, N., & Mandai, M. Tumor Immune Microenvironment during Epithelial-Mesenchymal Transition. Clinical cancer research 2021; 27(17), 4669–4679. https://doi.org/10.1158/1078-0432.CCR-20-4459
- Takeichi M. Cadherins in cancer: implications for invasion and metastasis. Current opinion in cell biology, 1993; 5(5), 806–811. https://doi.org/10.1016/0955-0674(93)90029-p
- Cowell, C. F., Yan, I. K., Eiseler, T., Leightner, A. C., Döppler, H., & Storz, P. Loss of cell-cell contacts induces NF-kappaB via RhoAmediated activation of protein kinase D1. Journal of cellular biochemistry, 2009; 106(4), 714–728. https://doi.org/10.1002/jcb.22067
- Hoesel, B., & Schmid, J. A. The complexity of NF-κB signaling in inflammation and cancer. Molecular Cancer 2013; 12(1), 86. doi:10.1186/1476-4598-12-86
- Ben-Neriah, Y., & Karin, M. Inflammation meets cancer, with NFκB as the matchmaker. Nature immunology 2011; 12(8), 715–723. https://doi.org/10.1038/ni.2060
- Sokolova, O., & Naumann, M. NF-κB Signaling in Gastric Cancer. Toxins 2017; 9(4), 119. https://doi.org/10.3390/toxins9040119
- Kuphal, S., Poser, I., Jobin, C., Hellerbrand, C., & Bosserhoff, A. K. Loss of E-cadherin leads to upregulation of NFkappaB activity in malignant melanoma. Oncogene 2004; 23(52), 8509–8519. https://doi.org/10.1038/sj.onc.1207831.
- Lin, T. C., Yang, C. H., Cheng, L. H., Chang, W. T., Lin, Y. R., & Cheng, H. C. Fibronectin in Cancer: Friend or Foe. Cells, 2019; 9(1), 27. https://doi.org/10.3390/cells9010027
- Sheng, S., Guo, B., Wang, Z., Zhang, Z., Zhou, J., & Huo, Z. Aberrant Methylation and Immune Microenvironment Are Associated With Overexpressed Fibronectin 1: A Diagnostic and Prognostic Target in Head and Neck Squamous Cell Carcinoma. Frontiers in molecular biosciences 2021; 8, 753563. https://doi.org/10.3389/fmolb.2021.753563
- Lamouille, S., Xu, J., & Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews. Molecular cell biology 2014; 15(3), 178–196. https://doi.org/10.1038/nrm3758
- Topalovski, M., & Brekken, R. A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer letters, 2016; 381(1), 252–258. https://doi.org/10.1016/j.canlet.2015.12.027
- Kuonen, F., Surbeck, I., Sarin, K. Y., Dontenwill, M., Rüegg, C., Gilliet, M., Oro, A. E., & Gaide, O. TGFβ, Fibronectin and Integrin α5β1 Promote Invasion in Basal Cell Carcinoma. The Journal of investigative dermatology 2018; 138(11), 2432–2442. https://doi.org/10.1016/j.jid.2018.04.029
- Takei, H., Iino, Y., Horiguchi, J., & Yokoe, T. Immunohistochemical fibronectin staining pattern and prognosis in invasive breast carcinoma. Oncology 1995;52(2), 106–111. https://doi.org/10.1159/000227439
- Jagirdar, J., Ishak, K. G., Colombo, M., Brambilla, C., & Paronetto, F. Fibronectin patterns in hepatocellular carcinoma and its clinical significance. Cancer, 1985; 56(7), 1643–1648. https://doi.org/10.1002/1097-0142(19851001)56:7<1643: aidcncr2820560730>3.0.co;2-o
- Stenman, S., & Vaheri, A. Fibronectin in human solid tumors. International journal of cancer, 1981; 27(4), 427–435. https://doi.org/10.1002/ijc.2910270403
- Grither, W. R., Divine, L. M., Meller, E. H., Wilke, D. J., Desai, R. A., et al. TWIST1 induces expression of discoidin domain receptor 2 to promote ovarian cancer metastasis. Oncogene 2018; 37(13), 1714–1729. https://doi.org/10.1038/s41388-017-0043-9
- Glasner, A., Levi, A., Enk, J., Isaacson, B., Viukov, S., Orlanski, S., Mandelboim, NKp46 Receptor-Mediated Interferon-γ Production by Natural Killer Cells Increases Fibronectin 1 to Alter Tumor Architecture and Control Metastasis. Immunity 2018; 48(1), 107–119.e4. doi: 10.1016/j.immuni.2017.12.007
- Omigbodun, A., Coukos, G., Ziolkiewicz, P., Wang, C. L., & Coutifaris, C. Macrophage-colony stimulating factor (M-CSF) regulates the expression of fibronectin and its alpha5 integrin receptor in human trophoblasts. Endocrinology, 1998; 139(4), 2190–2193. https://doi.org/10.1210/endo.139.4.6031
- Huang, L., Xu, X., & Hao, Y. The possible mechanisms of tumor progression via CSF-1/CSF-1R pathway activation. Romanian journal of morphology and embryology 2014;55(2 Suppl), 501–506.
- Ray, S., Ju, X., Sun, H., Finnerty, C. C., Herndon, D. N., & Brasier, A. R. The IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. The Journal of investigative dermatology 2013; 133(5), 1212–1220. https://doi.org/10.1038/jid.2012.499
- Zhang, F., Li, C., Halfter, H., & Liu, J. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene, 2003; 22(6), 894–905. https://doi.org/10.1038/sj.onc.1206158
- Ray, S., Ju, X., Sun, H., Finnerty, C. C., Herndon, D. N., & Brasier, A. R. (2013). The IL-6 trans-signaling-STAT3 pathway mediates ECM and cellular proliferation in fibroblasts from hypertrophic scar. The Journal of investigative dermatology, 133(5), 1212–1220. https://doi.org/10.1038/jid.2012.499
- Zhang, F., Li, C., Halfter, H., & Liu, J. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene 2003; 22(6), 894–905. https://doi.org/10.1038/sj.onc.1206158
- Han, Y., Liu, D., & Li, L. PD-1/PD-L1 pathway: current researches in cancer. American journal of cancer research, 2020 10(3), 727–742.
- Hou, P., Li, L., Chen, F., Chen, Y., Liu, H., Li, J., Bai, J., & Zheng, J. PTBP3-Mediated Regulation of ZEB1 mRNA Stability Promotes Epithelial-Mesenchymal Transition in Breast Cancer. Cancer research 2018;78(2), 387–398. https://doi.org/10.1158/0008-5472.CAN-17-0883
- Perdigão-Henriques, R., Petrocca, F., Altschuler, G., Thomas, M. P., Le, M. T., Tan, S. M., Hide, W., & Lieberman, J. miR-200 promotes the mesenchymal to epithelial transition by suppressing multiple members of the Zeb2 and Snail1 transcriptional repressor complexes. Oncogene, 2016; 35(2), 158–172. https://doi.org/10.1038/onc.2015.69
- Kun-Peng, Z., Chun-Lin, Z., Xiao-Long, M., & Lei, Z. Fibronectin-1 modulated by the long noncoding RNA OIP5-AS1/miR-200b-3p axis contributes to doxorubicin resistance of osteosarcoma cells. Journal of cellular physiology 2019; 234(5), 6927–6939. https://doi.org/10.1002/jcp.27435
- Ocaña, O. H., Córcoles, R., Fabra, A., Moreno-Bueno, G., Acloque, H., Vega, S., Barrallo-Gimeno, A., Cano, A., & Nieto, M. A. Metastatic colonization requires the repression of the epithelialmesenchymal transition inducer Prrx1. Cancer cell, 2012; 22(6), 709–724. https://doi.org/10.1016/j.ccr.2012.10.012
- Bradshaw, A. D., & Sage, E. H. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. The Journal of clinical investigation, 2001; 107(9), 1049–1054. https://doi.org/10.1172/JCI12939
- Tang, M. J., & Tai, I. T. A novel interaction between procaspase 8 and SPARC enhances apoptosis and potentiates chemotherapy sensitivity in colorectal cancers. The Journal of biological chemistry, 2007; 282(47), 34457–34467. https://doi.org/10.1074/jbc.M704459200
- Bradshaw, A. D., & Sage, E. H. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. The Journal of clinical investigation, 2001; 107(9), 1049–1054. https://doi.org/10.1172/JCI12939
- Nagaraju, G. P., Dontula, R., El-Rayes, B. F., & Lakka, S. S. Molecular mechanisms underlying the divergent roles of SPARC in human carcinogenesis. Carcinogenesis 2014; 35(5), 967–973. https://doi.org/10.1093/carcin/bgu072
- Brekken, R. A., & Sage, E. H.. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix biology 2001; 19(8), 816–827. https://doi.org/10.1016/s0945-053x(00)00133-5
- Nagaraju, G. P., Dontula, R., El-Rayes, B. F., & Lakka, S. S. Molecular mechanisms underlying the divergent roles of SPARC in human carcinogenesis. Carcinogenesis 2014; 35(5), 967–973. https://doi.org/10.1093/carcin/bgu072
- Stanisavljevic, J., Porta-de-la-Riva, M., Batlle, R., de Herreros, A. G., & Baulida, J. The p65 subunit of NF-κB and PARP1 assist Snail1 in activating fibronectin transcription. Journal of cell science, 2011; 124(Pt 24), 4161–4171. https://doi.org/10.1242/jcs.078824
- Sponziello, M., Rosignolo, F., Celano, M., Maggisano, V., Pecce, V., De Rose, R. F., Lombardo, G. E., Durante, C., Filetti, S., Damante, G., Russo, D., & Bulotta, S. Fibronectin-1 expression is increased in aggressive thyroid cancer and favors the migration and invasion of cancer cells. Molecular and cellular endocrinology, 2016; 431, 123–132. https://doi.org/10.1016/j.mce.2016.05.007
- Said N. Roles of SPARC in urothelial carcinogenesis, progression and metastasis. Oncotarget 2016; 7:67574–67585. doi: 10.18632/oncotarget.11590.
- Helene Sage, E. Terms of attachment: SPARC and tumorigenesis. Nature Medicine, 1997; 3(2), 144–146. doi:10.1038/nm0297-144 S
- Porter, P. L., Sage, E. H., Lane, T. F., Funk, S. E., & Gown, A. M. Distribution of SPARC in normal and neoplastic human tissue. The journal of histochemistry and cytochemistry 1995; 43(8), 791–800. https://doi.org/10.1177/43.8.7622842
- Massi, D., Franchi, A., Borgognoni, L., Reali, U. M., & Santucci, M. Osteonectin expression correlates with clinical outcome in thin cutaneous malignant melanomas. Human pathology 1999; 30(3), 339–344. https://doi.org/10.1016/s0046-8177(99)90014-x
- Chlenski, A., Liu, S., Crawford, S. E., Volpert, O. V., DeVries, G. H., Evangelista, A., et al. SPARC is a key Schwannian-derived inhibitor controlling neuroblastoma tumor angiogenesis. Cancer research, 2002; 62(24), 7357–7363.
- Zhang, Y., Yang, B., Du, Z., Bai, T., Gao, Y. T., Wang, Y. J., Lou, C., Wang, F. M., & Bai, Y. Aberrant methylation of SPARC in human hepatocellular carcinoma and its clinical implication. World journal of gastroenterology 2012; 18(17), 2043–2052. https://doi.org/10.3748/wjg.v18.i17.2043
- Liang, J. F., Wang, H. K., Xiao, H., Li, N., Cheng, C. X., Zhao, Y. Z., Ma, Y. B., Gao, J. Z., Bai, R. B., & Zheng, H. X. Relationship and prognostic significance of SPARC and VEGF protein expression in colon cancer. Journal of experimental & clinical cancer research: CR, 2010;29(1), 71. https://doi.org/10.1186/1756-9966-29-71
- Socha, M. J., Said, N., Dai, Y., Kwong, J., Ramalingam, P., Trieu, V., Desai, N., Mok, S. C., & Motamed, K. Aberrant promoter methylation of SPARC in ovarian cancer. Neoplasia 2009; 11(2), 126–135. https://doi.org/10.1593/neo.81146
- Thomas, R., True, L. D., Bassuk, J. A., Lange, P. H., & Vessella, R. L. Differential expression of osteonectin/SPARC during human prostate cancer progression. Clinical cancer 2000; 6(3), 1140–1149.
- Gao, J., Song, J., Huang, H., Li, Z., Du, Y., Cao, J., Li, M., Lv, S., Lin, H., & Gong, Y. Methylation of the SPARC gene promoter and its clinical implication in pancreatic cancer. Journal of experimental & clinical cancer research: CR, 2010; 29(1), 28. https://doi.org/10.1186/1756-9966-29-28
- Guweidhi, A., Kleeff, J., Adwan, H., Giese, N. A., Wente, M. N., Giese, T., Büchler, M. W., Berger, M. R., & Friess, H. Osteonectin influences growth and invasion of pancreatic cancer cells. Annals of surgery, 2005; 242(2), 224–234. https://doi.org/10.1097/01.sla.0000171866.45848.68
- Chen, G., Tian, X., Liu, Z., Zhou, S., Schmidt, B., Henne-Bruns, D., Bachem, M., & Kornmann, M. Inhibition of endogenous SPARC enhances pancreatic cancer cell growth: modulation by FGFR1-III isoform expression. British journal of cancer, 2010; 102(1), 188–195. https://doi.org/10.1038/sj.bjc.6605440
- Inoue, M., Senju, S., Hirata, S., Ikuta, Y., Hayashida, Y., Irie, A., Harao, M., Imai, K., Tomita, Y., Tsunoda, T., Furukawa, Y., Ito, T., Nakamura, Y., Baba, H., & Nishimura, Y. Identification of SPARC as a candidate target antigen for immunotherapy of various cancers. International journal of cancer 2010; 127(6), 1393–1403. https://doi.org/10.1002/ijc.25160
- Hu, J., Ma, Y., Ma, J., Chen, S., Zhang, X., Guo, S., Huang, Z., Yue, T., Yang, Y., Ning, Y., Zhu, J., Wang, P., Wang, X., Chen, G., & Liu, Y. Macrophage-derived SPARC Attenuates M2-mediated Pro-tumour Phenotypes. Journal of Cancer, 2020;11(10), 2981–2992. https://doi.org/10.7150/jca.39651
- Wang, L., Wang, W., Xu, Y., & Wang, Q. Low Levels of SPARC are Associated with Tumor Progression and Poor Prognosis in Human Endometrial Carcinoma. OncoTargets and therapy 2020; 13, 11549–11569. https://doi.org/10.2147/OTT.S277795
- Liang, J. F., Wang, H. K., Xiao, H., Li, N., Cheng, C. X., Zhao, Y. Z., Ma, Y. B., Gao, J. Z., Bai, R. B., & Zheng, H. X. Relationship and prognostic significance of SPARC and VEGF protein expression in colon cancer. Journal of experimental & clinical cancer research: CR, 2010 29(1), 71. https://doi.org/10.1186/1756-9966-29-71
- Villarejo, A., Cortés-Cabrera, A., Molina-Ortíz, P., Portillo, F., & Cano, A. Differential role of Snail1 and Snail2 zinc fingers in Ecadherin repression and epithelial to mesenchymal transition. The Journal of biological chemistry, 2014; 289(2), 930–941. https://doi.org/10.1074/jbc.M113.528026
- Wong, S. L., & Sukkar, M. B. The SPARC protein: an overview of its role in lung cancer and pulmonary fibrosis and its potential role in chronic airways disease. British journal of pharmacology, 2017; 174(1), 3–14. https://doi.org/10.1111/bph.13653
- Grant JL, Fishbein MC, Hong LS, Krysan K, Minna JD, Shay JW et al. A novel molecular pathway for snail-dependent, SPARCmediated invasion in non-small cell lung cancer pathogenesis. Cancer Prev Res 2014; 7: 150–160.
- Vaz, J., Ansari, D., Sasor, A., & Andersson, R. SPARC: A Potential Prognostic and Therapeutic Target in Pancreatic Cancer. Pancreas 2015; 44(7), 1024–1035. https://doi.org/10.1097/MPA.0000000000000409
- Shawar, S. M., Vyas, J. M., Rodgers, J. R., & Rich, R. R. Antigen presentation by major histocompatibility complex class I-B molecules. Annual review of immunology 1994; 12, 839–880. https://doi.org/10.1146/annurev.iy.12.040194.004203
- Xu, X., Zhou, Y., & Wei, H. Roles of HLA-G in the Maternal-Fetal Immune Microenvironment. Frontiers in immunology, 2020; 11, 592010. https://doi.org/10.3389/fimmu.2020.592010
- Van Hall, T., Oliveira, C. C., Joosten, S. A., & Ottenhoff, T. H. M. The other Janus face of Qa-1 and HLA-E: diverse peptide repertoires in times of stress. Microbes and Infection, 2010; 12(12-13), 910–918. doi: 10.1016/j.micinf.2010.07.011
- Sullivan, L. C., Clements, C. S., Rossjohn, J., & Brooks, A. G. The major histocompatibility complex class Ib molecule HLA-E at the interface between innate and adaptive immunity. Tissue antigens 2008; 72(5), 415–424. https://doi.org/10.1111/j.1399-0039.2008.01138.x
- Wada, H., Matsumoto, N., Maenaka, K., Suzuki, K., & Yamamoto, K. The inhibitory NK cell receptor CD94/NKG2A and the activating receptor CD94/NKG2C bind the top of HLA-E through mostly shared but partly distinct sets of HLA-E residues. European journal of immunology, 2004;34(1), 81–90. https://doi.org/10.1002/eji.200324432.
- Kaiser, B. K., Barahmand-Pour, F., Paulsene, W., Medley, S., Geraghty, D. E., & Strong, R. K. Interactions between NKG2x immunoreceptors and HLA-E ligands display overlapping affinities and thermodynamics. Journal of immunology 2005; 174(5), 2878–2884. https://doi.org/10.4049/jimmunol.174.5.2878
- Valés-Gómez, M., Reyburn, H. T., Erskine, R. A., López-Botet, M., & Strominger, J. L. Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. The EMBO journal, 1999; 18(15), 4250–4260. https://doi.org/10.1093/emboj/18.15.4250
- Borst, L., van der Burg, S. H., & van Hall, T. The NKG2A-HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clinical cancer research, 2020; 26(21), 5549–5556.
- Wieten, L., Mahaweni, N. M., Voorter, C. E. M., Bos, G. M. J., & Tilanus, M. G. J. Clinical and immunological significance of HLA-E in stem cell transplantation and cancer. Tissue Antigens, 2014; 84(6), 523–535. doi:10.1111/tan.12478
- Gooden, M., Lampen, M., Jordanova, E. S., Leffers, N., Trimbos, J. B., van der Burg, S. H., Nijman, H., & van Hall, T. (). HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8⁺ T lymphocytes. Proceedings of the National Academy of Sciences of the United States of America, 2011; 108(26), 10656–10661. https://doi.org/10.1073/pnas.1100354108
- Seliger, B., Jasinski-Bergner, S., Quandt, D., Stoehr, C., Bukur, J., Wach, S., Legal, W., Taubert, H., Wullich, B., & Hartmann, A. HLA-E expression and its clinical relevance in human renal cell carcinoma. Oncotarget 2016; 7(41), 67360–67372. https://doi.org/10.18632/oncotarget.11744
- Levy, E. M., Bianchini, M., Von Euw, E. M., Barrio, M. M., Bravo, A. I., Furman, D., Domenichini, E. et al. Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. International journal of oncology 2008; 32(3), 633–641.
- Borst, L., van der Burg, S. H., & van Hall, T. The NKG2A-HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clinical cancer research 2020; 26(21), 5549–5556. https://doi.org/10.1158/1078-0432.CCR-19-2095
- 167.- Andersson, E., Poschke, I., Villabona, L., Carlson, J. W., Lundqvist, A., Kiessling, R., Seliger, B., & Masucci, G. V. Non-classical HLA-class I expression in serous ovarian carcinoma: Correlation with the HLA-genotype, tumor infiltrating immune cells and prognosis. Oncoimmunology, 2015; 5(1), e1052213. https://doi.org/10.1080/2162402X.2015.1052213
- de Kruijf, E. M., Sajet, A., van Nes, J. G., Natanov, R., Putter, H., Smit, V. T., Liefers, G. J., van den Elsen, P. J., van de Velde, C. J., & Kuppen, P. J. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. Journal of immunology 2010 185(12), 7452–7459. https://doi.org/10.4049/jimmunol.1002629
- Gooden, M., Lampen, M., Jordanova, E. S., Leffers, N., Trimbos, J. B., van der Burg, S. H., Nijman, H., & van Hall, T. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8⁺ T lymphocytes. Proceedings of the National Academy of Sciences of the United States of America, 201; 108(26), 10656–10661. https://doi.org/10.1073/pnas.1100354108
- van Esch, E. M., Tummers, B., Baartmans, V., Osse, E. M., Ter Haar, N., Trietsch, M. D., Hellebrekers, B. W., Holleboom, C. A., Nagel, H. T., Tan, L. T., Fleuren, G. J., van Poelgeest, M. I., van der Burg, S. H., & Jordanova, E. S. Alterations in classical and nonclassical HLA expression in recurrent and progressive HPV-induced usual vulvar intraepithelial neoplasia and implications for immunotherapy. International journal of cancer, 2014;135(4), 830–842. https://doi.org/10.1002/ijc.28713
- Talebian Yazdi, M., van Riet, S., van Schadewijk, A., Fiocco, M., van Hall, T., Taube, C., Hiemstra, P. S., & van der Burg, S. H. The positive prognostic effect of stromal CD8+ tumor-infiltrating T cells is restrained by the expression of HLA-E in non-small cell lung carcinoma. Oncotarget, 2016; 7(3), 3477–3488. https://doi.org/10.18632/oncotarget.6506
- Lo, S. H., & Lo, T. B. Cten, a COOH-terminal tensin-like protein with prostate restricted expression, is down-regulated in prostate cancer. Cancer research 2002 62(15), 4217–4221.
- Lo S. H. C-terminal tensin-like (CTEN): a promising biomarker and target for cancer. The international journal of biochemistry & cell biology, 2014; 51, 150–154. https://doi.org/10.1016/j.biocel.2014.04.003
- Lo, S. H., Weisberg, E., & Chen, L. B. Tensin: a potential link between the cytoskeleton and signal transduction. BioEssays: news and reviews in molecular, cellular and developmental biology, 1994;16(11), 817–823. https://doi.org/10.1002/bies.950161108
- Lo, S. H., Janmey, P. A., Hartwig, J. H., & Chen, L. B. Interactions of tensin with actin and identification of its three distinct actin-binding domains. The Journal of cell biology, 1994; 125(5), 1067–1075. https://doi.org/10.1083/jcb.125.5.1067
- Katz, M., Amit, I., Citri, A., Shay, T., Carvalho, S., Lavi, S., et al. A reciprocal tensin-3–cten switch mediates EGF-driven mammary cell migration. Nature Cell Biology 2007; 9(8), 961–969. doi:10.1038/ncb1622.
- Liao, Y. C., Chen, N. T., Shih, Y. P., Dong, Y., & Lo, S. H. Up-regulation of C-terminal tensin-like molecule promotes the tumorigenicity of colon cancer through beta-catenin. Cancer research, 2009; 69(11), 4563–4566. https://doi.org/10.1158/0008-5472.CAN-09-0117
- Sasaki H, Moriyama S, Mizuno K, Yukiue H, Konishi A, Yano M, Kaji M, Fukai I, Kiriyama M, Yamakawa Y, Fujii Y. CTEN mRNA expression was correlated with tumor progression in lung cancers. Lung Cancer. 2003; 40:151–155
- Liao, Y. C., Chen, N. T., Shih, Y. P., Dong, Y., & Lo, S. H. Up-regulation of C-terminal tensin-like molecule promotes the tumorigenicity of colon cancer through beta-catenin. Cancer research 2009; 69(11), 4563–4566. https://doi.org/10.1158/0008-5472.CAN-09-0117
- Sakashita, K., Mimori, K., Tanaka, F., Kamohara, Y., Inoue, H., Sawada, T., Hirakawa, K., & Mori, M. Prognostic relevance of Tensin4 expression in human gastric cancer. Annals of surgical oncology, 2008; 15(9), 2606–2613. https://doi.org/10.1245/s10434-008-9989-8
- Albasri, A., Aleskandarany, M., Benhasouna, A., Powe, D. G., Ellis, I. O., Ilyas, M., & Green, A. R. CTEN (C-terminal tensin-like), a novel oncogene overexpressed in invasive breast carcinoma of poor prognosis. Breast cancer research and treatment, 2011; 126(1), 47–54. https://doi.org/10.1007/s10549-010-0890-3
- Li, Y., Mizokami, A., Izumi, K., Narimoto, K., Shima, T., Zhang, J., Dai, J., Keller, E. T., & Namiki, M. CTEN/tensin 4 expression induces sensitivity to paclitaxel in prostate cancer. The Prostate, 2010; 70(1), 48–60. https://doi.org/10.1002/pros.21037
- Al-Ghamdi, S., Albasri, A., Cachat, J., Ibrahem, S., Muhammad, B. A., Jackson, D., Ilyas, M. Cten Is Targeted by Kras Signalling to Regulate Cell Motility in the Colon and Pancreas. PLoS ONE2011; 6(6), e20919. doi: 10.1371/journal.pone.0020919
- Wu, W. M., & Liao, Y. C. Downregulation of C-Terminal Tensin-Like Protein (CTEN) Suppresses Prostate Cell Proliferation and Contributes to Acinar Morphogenesis. International journal of molecular sciences,2018; 19(10), 3190. https://doi.org/10.3390/ijms19103190
- Shivanna, S., Harrold, I., Shashar, M., Meyer, R., Kiang, C., Francis, J., Zhao, Q., Feng, H., Edelman, E. R., Rahimi, N., & Chitalia, V. C. The c-Cbl ubiquitin ligase regulates nuclear β-catenin and angiogenesis by its tyrosine phosphorylation mediated through the Wnt signaling pathway. The Journal of biological chemistry, 2015¸ 290(20), 12537–12546. https://doi.org/10.1074/jbc.M114.616623
- Silva-García, O., Valdez-Alarcón, J. J., & Baizabal-Aguirre, V. M. Wnt/β-Catenin Signaling as a Molecular Target by Pathogenic Bacteria. Frontiers in immunology, 2019;10, 2135. https://doi.org/10.3389/fimmu.2019.02135
- Easwaran, V., Lee, S. H., Inge, L., Guo, L., Goldbeck, C., Garrett, E., Wiesmann, M., Garcia, P. D., Fuller, J. H., Chan, V., et al. betaCatenin regulates vascular endothelial growth factor expression in colon cancer. Cancer research, 2003; 63(12), 3145–3153.
- Lu, X., Zhou, B., Cao, M., Shao, Q., Pan, Y., & Zhao, T. CTEN Inhibits Tumor Angiogenesis and Growth by Targeting VEGFA Through Down-Regulation of β-Catenin in Breast Cancer. Technology in cancer research & treatment, 2021; 20, 15330338211045506. https://doi.org/10.1177/15330338211045506
- Albasri, A., Aleskandarany, M., Benhasouna, A., Powe, D. G., Ellis, I. O., Ilyas, M., & Green, A. R. CTEN (C-terminal tensin-like), a novel oncogene overexpressed in invasive breast carcinoma of poor prognosis. Breast cancer research and treatment, 2011; 126(1), 47–54. https://doi.org/10.1007/s10549-010-0890-3
- Barbieri I, Pensa S, Pannellini T, Quaglino E, Maritano D, Demaria M, et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res 2010; 70: 2558–2567. https://doi.org/10.1158/0008-5472.
- Albasri A, Seth R, Jackson D, Benhasouna A, Crook S, Nateri AS, et al. C-terminal Tensin-like (CTEN) is an oncogene which alters cell motility possibly through repression of E-cadherin in colorectal cancer. J Pathol 2009;218: 57–65. https://doi.org/10.1002/path.2508 PMID: 19214987.
- Lu X, Gao J, Zhang Y, Zhao T, Cai H, Zhang T. CTEN induces epithelialmesenchymal transition (EMT) and metastasis in non small cell lung cancer cells. PLoS ONE 2018; 13(7): e0198823. https://doi.org/10.1371/journal. pone.0198823.
- Qi, X., Sun, L., Wan, J., Xu, R., He, S., & Zhu, X. Tensin4 promotes invasion and migration of gastric cancer cells via regulating AKT/GSK-3β/snail signaling pathway. Pathology - Research and Practice, 2020; 216(7), 153001. doi: 10.1016/j.prp.2020.153001
- Bai, Q., Song, D., Gu, L., Verkhratsky, A., & Peng, L. Bi-phasic regulation of glycogen content in astrocytes via Cav-1/PTEN/PI3K/AKT/GSK-3β pathway by fluoxetine. Psychopharmacology, 2017; 234(7), 1069–1077. https://doi.org/10.1007/s00213-017-4547-3
- Zhou, S. L., Zhou, Z. J., Hu, Z. Q., Li, X., Huang, X. W., Wang, Z., Fan, J., Dai, Z., & Zhou, J. CXCR2/CXCL5 axis contributes to epithelial-mesenchymal transition of HCC cells through activating PI3K/Akt/GSK-3β/Snail signaling. Cancer letters, 2015; 358(2), 124–135. https://doi.org/10.1016/j.canlet.2014.11.044S
- K. Aratani, S. Komatsu, D. Ichikawa, T. Ohashi, M. Miyamae, W. Okajima, T. Imamura, J. Kiuchi, K.et al. Overexpression of CTEN relates to tumor malignant potential and poor outcomes of adenocarcinoma of the esophagogastric junction, Oncotarget 2017; 8 84112–84122.
- Siewert, J. R., Böttcher, K., Stein, H. J., & Roder, J. D. Relevant prognostic factors in gastric cancer: ten-year results of the German Gastric Cancer Study. Annals of surgery, 1998; 228(4), 449–461. https://doi.org/10.1097/00000658-199810000-00002