
Japanese Journal of Gastroenterology Research
Research Article - Open Access, Volume 2
Analysis of MLH1 promoter methylation by different methods in colorectal tumors
Marcelo Vital1,2; Carolina Vergara3; Florencia Carusso3; Florencia Neffa3; Adriana Della Valle3; Patricia Esperón2,3,4*
1Graduate Program in Chemistry, Facultad de Química, Universidad de la República, Uruguay.
2Molecular Genetics Unit, School of Chemistry, Universidad de la República, Montevideo, Uruguay
3Uruguayan Collaborative Group, Dirección Nacional de Sanidad de las FFAA, Montevideo, Uruguay.
4Latin American Network for Implementation and Validation of Clinical Pharmacogenomics Guidelines (RELIVAF-CYTED), Madrid, Spain.
*Corresponding Author : Patricia Esperón
Molecular Genetics Unit, School of Chemistry, Universidad de la República, Montevideo, Uruguay.
Email: pesperon@fq.edu.uy
Received : Nov 04, 2022
Accepted : Dec 07, 2022
Published : Dec 14, 2022
Archived : www.jjgastro.com
Copyright : © Esperón P (2022).
Abstract
Colorectal Cancer (CRC) is the second leading cause of cancerrelated death worldwide, affecting both men and women. A genetic CRC characteristic is the presence of microsatellite instability (MSI). This instability could result from the inactivation of the Mismatch Repair (MMR) system by either MMR gene mutations, or MMR gene promoter hypermethylation, mainly in the MLH1 gene promoter. Lynch Syndrome (LS), the most common form of hereditary colorectal cancer, is caused by germline mutations in MMR genes. When a tumor has a high MSI, prior to the germline genetic study of Lynch Syndrome, a methylation study is recommended to rule out the diagnosis of sporadic colorectal cancer. The MLH1 gene promoter methylation status of sixty-three MSI colon tumors and ten normal colon tissues using different techniques was evaluated. DNA sequencing, Real-Time PCR-High Resolution Melting (PCR-HRM), methylationspecific-PCR, Combined Bisulphite Restriction Analysis (COBRA) and Methylation Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA) techniques were tested. The same ten MSI samples were found to be methylated using any of the four techniques. Since all the methods similarly evaluated the MLH1 methylation status, which one to choose would depend on the convenience and availability of each laboratory. After our evaluation based on the main advantages (short turnaround time, high throughput, sensitivity, and ease of performance), we concluded that PCR-HRM was the most convenient screening method for the MLH1 methylation study.
Keywords: Colorectal cancer; MLH1 promoter.
Citation: Vital M, Vergara C, Carusso F, Neffa F, Valle AD, et al. Analysis of MLH1 promoter methylation by different methods in colorectal tumors. Japanese J Gastroenterol Res. 2022; 2(15): 1123.
Introduction
Colorectal Cancer (CRC) is the second leading cause of cancer-related death worldwide, affecting both men and women [1,2]. Several risk factors are associated with an increased risk for CRC, some are modifiable (e.g., obesity; diet; long-term smoking), others hereditary (e.g. Lynch Syndrome or familial adenomatous polyposis) [3].
CRC results from the progressive accumulation over time of morphological, genetic and epigenetic changes that lead to the transformation from normal colonic epithelial cells to colon adenocarcinoma cells [4]. Some pathways can contribute to the carcinogenic process, e.g., Chromosomal Instability (CIN), microsatellite instability (MSI) or CpG island methylator phenotype (CIMP) [5]. CIN is the most common CRC pathway and consists of karyotypic abnormalities and an accumulation of a characteristic set of mutations in specific tumour suppressor genes and oncogenes [6]. MSI is a result of genetic instability in short nucleotide repeats that occurs spontaneously during DNA replication due to a defective DNA mismatch repair (MMR) system. Lynch Syndrome (LS), the most common form of hereditary CRC (3-5%), is mainly due to germline mutations in MMR genes (MLH1, MSH2, MSH6, and PMS2) and more than 90% of tumours are MSI-High (MSI-H) [7,8]. The CIMP phenotype is due to gene promoter hypermethylation in CpG dinucleotide regions causing gene expression silencing. The bulk of MSI-H tumours are sporadic and only 15% have a hereditary origin.
10-15% of sporadic CRCs shows MSI-H being mainly due to epigenetic silencing of MLH1 gene [9]. The methylation of regions C and D located in MLH1 gene promoter (figure 1) was correlated to the absence of protein expression [10,11].
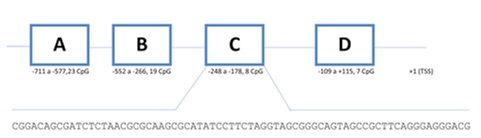
A large number of DNA methylation profiling techniques have been described. Most are PCR-based techniques, such as methylation-specific-PCR (MS-PCR) [9], real-time-PCR [12], Combined Bisulphite Restriction Analysis (COBRA) [13] or Methylation Specific Multiplex Ligation-dependent Probe Amplification (MS-MLPA) [14]. Bisulphite DNA sequencing method developed by Clark et al [15], is considered the gold standard for methylation analysis [16]. Most methods include a DNA modification treatment with sodium bisulphite before any further amplification round.
The aim of this study was to evaluate the performance of five methods (DNA sequencing, PCR-HRM, MS-PCR, COBRA and MS-MLPA) for the analysis of MLH1 gene promoter methylation in MSI-High CRC tumours.
Materials and methods
Samples and DNA isolation
Tumors DNA from 63 colorectal cancer patients were included. All tumors were classified as MSI-H by PCR using the Bethesda reference panel of microsatellites. Tumors were defined as MSI-H when at least two or more markers were positive [17]. When formalin-fixed paraffin-embedded (FFPE) tissue samples were analyzed (42 samples), one or two 5µm sections were treated with xylene and ethanol before a proteinase K digestion, previous to the DNA purification using the QIAamp DNA FFPE Mini Kit (Qiagen, Valencia, CA, USA) following the manufacturer instructions (yield 10 to 40 ng/µL).
For the 21 frozen tissues tumors (starting amount <10mg), the PureLink™ Genomic DNA (Invitrogen, Carlsbad, CA, USA) was used (yield 70 to 120 ng/µL). DNA concentration was quantified using a NanoDrop 2000c spectrophotometer. A control group of 10 normal tissues was also analyzed.
This study was approved by the Institutional Bioethical Board. Study procedures were in accordance with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all individuals included in this study.
DNA bisulphite conversion
Bisulphite conversion was performed using the EZ DNA Methylation-Lightning Kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s protocol.
The sodium bisulphite treatment consists of a two-step reaction. First, the sulfonation at C-6 position of the cytosine produces a cytosine sulfonate that then became an uracil sulfonate by hydrolytic deamination. Finally, the removal of the sulfonate group, under alkaline conditions, leads to the conversion into uracil. If the CpG site is methylated, the methyl group at C-5 position blocks the sulphonate cytosine formation. In short, after the sodium bisulphite treatment, unmethylated cytosines converted into uracil, while methylated cytosines remain intact.
DNA sequencing
The bisulphite-modified DNA was amplified using two rounds of PCR with nested primers (table 1). The first PCR round was done in 1X PCR buffer, 0.2 μM of each primer, 0.8 nM of dNTPs, 1.5 mM of MgCl2, 1 μL of modified genomic DNA, and 0.3 U of Hot Start DNA polymerase (Promega) in a final volume of 10 μL. A second amplification with 1 uL of DNA obtained from the first round was carried out with the same reagents but with 2 mM of MgCl2 final concentration.
Table 1: PCR primers list. Primers used in bisulphite DNA sequencing, PCR-HRM and COBRA assays.
Sequence |
Length (bp) |
|
C- FE |
TAGGGTTAACGTTAGAAAGGTCGTAAGG |
544 |
C-RE |
ATAACATTAACTAACCGCTAAATAACTT |
|
C-FI |
AAAGGTCGTAAGGGGAGAGGAGGAGTT |
336 |
C-RI |
AACTTACGCCATCCAACCCCACCCTTC |
The thermal cycler program was set up as: 95oC for 5 minutes, 40 cycles of 95°C 30 sec, 52oC 30 sec, 72°C 30 sec, with a final extension for 5 min at 72°C.
PCR products were purified from 2% agarose gels using a commercial kit (QIAquick Gel Extraction Kit, Qiagen, Valencia, USA).
Purified fragments were sequenced using the BigDye® Terminator v3.1 Cycle Sequencing Kits (Applied Biosystems) and the ABI 3730xl DNA Analyzer (Applied Biosystems Inc., USA). Sequences were analyzed using BioEdit software (www.mbio.ncsu.edu/BioEdit/bioedit.html).
PCR-HRM
This technique was carried out in a Rotor Gene 6000 (Corbett Life Science, Qiagen, USA). The bisulphite-modified DNA was first amplified with primers C-FE and C-RE (Table 1). The second amplification round was set up with 0.7 μM of each primer [18], with 1 μL of DNA template obtained from the first round of PCR (544bp), in a master mix (Type IT HRM kit, Qiagen, Valencia, CA, USA). The optimized thermal cycling conditions included an initial hold at 95°C for 10 min, followed by 40 cycles of 95°C for 10s, 55°C for 15 s and 72°C for 10 s. The subsequent HRM analysis was performed with a continuous fluorescence acquisition mode from 73°C to 80°C at a ramp rate of 0.1°C/s.
MS-PCR
The bisulphite-modified DNA was amplified using primers specific for both methylated and unmethylated sequences [19]. Primers were designed with at least one CpG dinucleotide placed at the 3´ end to maximize the discrimination between both alleles. Fragments of 91bp or 102bp length were expected for the methylated or unmethylated alleles, respectively.
COBRA
After DNA amplification (see section 2.3) PCR products were digested with the BstUI restriction enzyme (New England Biolabs) that cleaves CGCG sites. Digested PCR products were analyzed on 6% polyacrylamide gels by silver nitrate staining.
MS-MLPA
The SALSA MS-MLPA Probemix ME011-C1 kit MMR genes (MRC-Holland, Amsterdam, The Netherlands) was performed as described by the manufacturer. 50-100 ng of genomic DNA was required. Fragments were analysed in an ABI 3500 System and the Coffalyser. Net software was used for data analysis.
All experiments were conducted in duplicate.
Results
Four different methods were employed to evaluate the MLH1 promoter methylation status of DNA obtained from both FFPE and frozen tissues.
After the DNA sequencing, the electropherogram showed full conversion of cytosine into thymine or an unchanged cytosine pattern at CpG sites from normal or tumour samples, respectively (Figure 2a). The PCR-HRM melting curves analysis showed different melting temperatures for methylated and unmethylated fragments, which allowed their discrimination (Figure 2b). Fragments that kept methylated cytosine, showed a greater melting temperature than unmethylated fragments in which cytosine was converted into thymine. The COBRA assay allowed fully differentiate DNA methylation since methylated fragments maintained the CGCG motif and were digested by BstUI enzyme (Figure 2c, lanes 4 and 5). MS-MLPA only amplified by PCR those fragments from methylated regions that could not be digested by the methylation-sensitive HhaI restriction enzyme (Figure 3). These last three methods were validated using methylated and non-methylated DNA samples, previously checked by DNA sequencing.
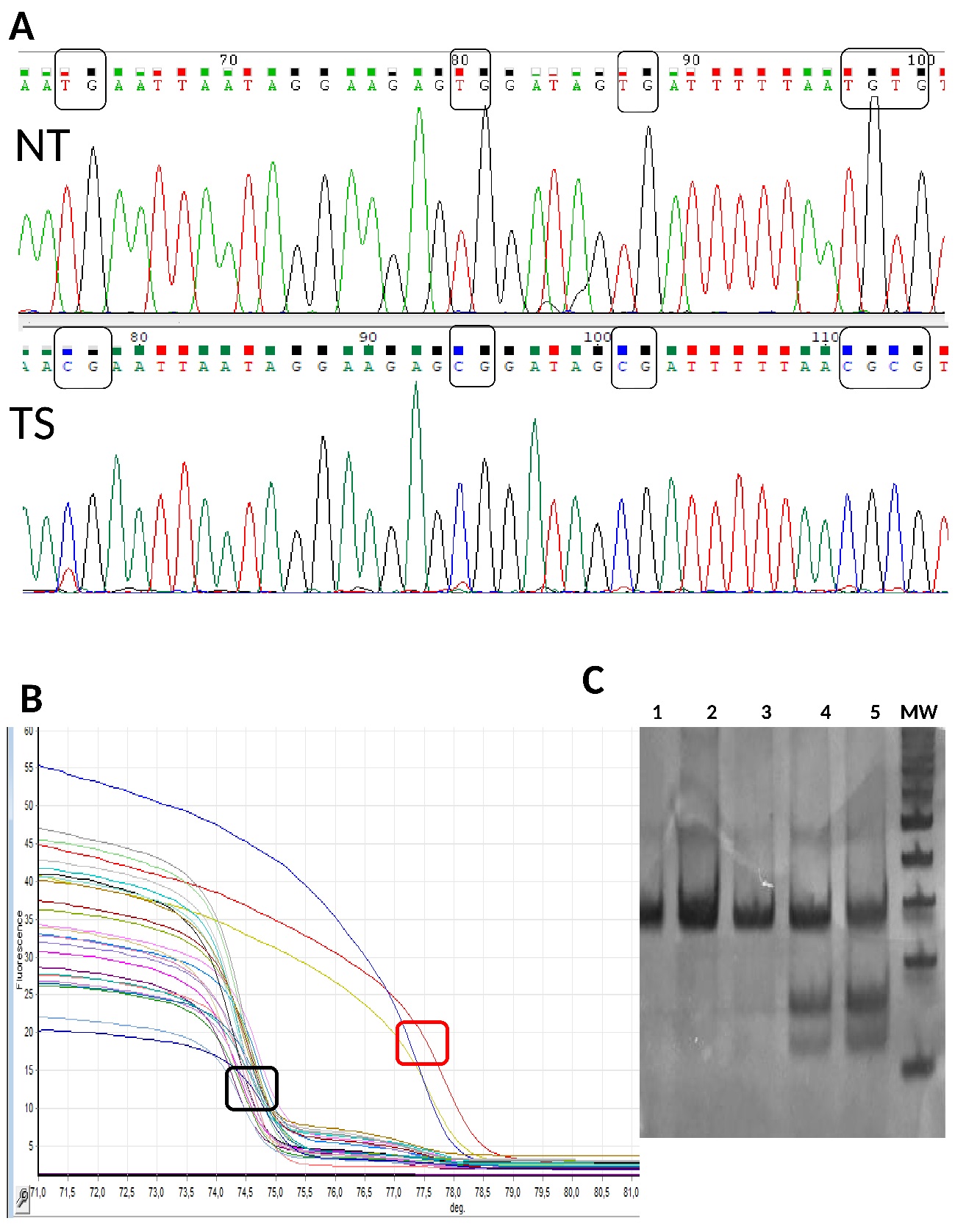
We found that methylated samples showed the expected profile curve at the PCR-HRM output, agreed with the methylation profile in the COBRA assay, and showed the predicted fragments in the MS-MLPA electropherograms.
These methods were performed successfully and harmonized results were obtained. Unfortunately, non-conclusive results were obtained with the MS-PCR technique.
In most cases (80%) biallelic methylation of MLH1 gene promoter was found. The heterozygosity pattern found in some samples is surely due to the coexistence of tumoral and normal cells.
Discussion
In this work, we evaluated the performance of DNA sequencing, PCR-HRM, COBRA, and MS-MLPA methods for the MLH1promoter methylation study of DNA samples obtained from both FFPE and frozen tissues.
A strength of this work was to obtain good-quality DNA from FFPE blocks that can subsequently be modified by bisulphite and amplified. Otherwise, DNA degradation would have made amplification difficult after bisulphite treatment.
The main advantage of DNA sequencing is that provides information on the methylation status of each CpG sites. However, like all sequencing technologies, the whole process requires a laborious protocol and a DNA sequencer. In addition, further bioinformatics analysis must be performed, that lead to a longer turnaround time.
COBRA is easily manipulated, only requires PCR reagents and equipment that can be found in any molecular biology laboratory.
Although MS-MLPA does not require sodium bisulphite treatment, a discrimination based on the methylation-sensitive HhaI enzyme is used. It involves multiple manipulation steps, a capillary electrophoresis device, and specific software for data analysis. Moreover, this assay evaluates the methylation profile of some other MMR genes, BRAFV600E among other gene variant analysis. The PCR-HRM assay can be implemented in resource-limited laboratories, and requires less laboratory working time than any of the techniques evaluated. Main advantages such as the absence of manual post-PCR treatment, a complete process in a closed-tube in a Real-Time thermal cycler, short turnaround time, high throughput, and quick analysis, make this assay, in our opinion, the most convenient screening method.
Conclusion
CRC tumours with MLH1 promoter hypermethylation have almost certainly a sporadic origin and excludes LS. Therefore, to determine the MLH1 methylation profile, a reliable technique must be performed. PCR-HRM, COBRA, MS-MLPA and DNA sequencing methods evaluated similarly the MLH1 methylation status. Then, which of the technique to choose will depend on the equipment and reagent availability, turnaround time, number of samples to be analysed, as well as staff laboratory skills.
References
- Bray F, Ferlay J, Soerjomataram I, Siegel R, Torre L, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2018; 68: 394-424.
- Vaccaro CA, Sarroca C, Rossi B, Lopez-Kostner F, Dominguez M, et al. Lynch syndrome in South America: past, present and future. Fam Cancer. 2016; 15: 437-445.
- American Cancer Society. Cancer Facts & Figures 2015. Atlanta, GA: American Cancer Society.
- Yamagishi H, Kuroda H, Imai Y, Hiraishi H. Molecular pathogenesis of sporadic colorectal cancers. Chin J Cancer. 2016; 35: 4.
- Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular Pathways Involved in Colorectal Cancer: Implications for Disease Behavior and Prevention. Int J Mol Sci. 2013; 14: 16365-16385.
- Pino N, Chung D. The chromosomal instability pathway in coloncancer. Gastroenterol. 2010; 138: 2059-2072.
- Duraturo F, Liccardo R, De Rosa M, Izzo P. Genetics, diagnosis and treatment of Lynch syndrome: Old lessons and current challenges. Oncol Lett. 2019; 17: 3048-3054.
- Bacher J, Sievers Ch, Albrecht D, Grimes I, Weiss J, et al. Improved Detection of Microsatellite Instability in Early Colorectal Lesions. PLoS ONE. 2015; 10: e0132727.
- Herman J, Umar A, Polyak K, Graff J, Ahuja N, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998; 95: 6870- 6875.
- Deng G, Chen A, Hong J, Chae H, Kim Y. Methylation of CpG in a Small Region of the hMLH1 Promoter Invariably Correlates with the Absence of Gene Expression. Cancer Res. 1999; 59: 2029- 2033.
- Deng G, Peng E, Gum J, Terdiman J, Sleisenger M, et al. Methylation of hMLH1 promoter correlates with the gene silencing with a region-specific manner in colorectal cancer. Br J Cancer. 2002; 86: 574-579.
- Eads CD, Danenberg K, Kawakami K, Saltz L, Blake C, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000; 28: E32.
- Xiong Z, Laird P. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997; 25: 2532-2534.
- Nygren A, Ameziane N, Duarte H, Vijzelaar R, Waisfisz Q, et al. Methylation-Specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res. 2005; 33: e128.
- Clark S, Harrison J, Paul C, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994; 22: 2990- 2997.
- Li Q, Hermanson P, Springer N. Detection of DNA methylation by whole-genome bisulphite sequencing. In Methods in Molecular Biology. Humana Press Inc. 2018; 185-196.
- Murphy KM, Zhang S, Geiger T, Hafez MJ, Bacher J, et al. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn. 2006; 8: 305-311.
- Gausachs M, Mur P, Corral J, González S, Benito L, et al. MLH1 promoter hypermethylation in the analytical algorithm of Lynch syndrome: a cost-effectiveness study. Eur J Hum Genet. 2012; 20: 762-768.
- Grady W, Rajput A, Lutterbaugh J, Markowitz S. Detection of Aberrantly Methylated hMLH1 Promoter DNA in the Serum of Patients with Microsatellite Unstable Colon Cancer. Cancer Res. 2001;61:900-902.