
Japanese Journal of Gastroenterology Research
Case Series - Open Access, Volume 2
Caroli’s disease in children: A case series
Galuh Martin Maytasari*; Titis Widowat
Department of Child Health, Faculty of Medicine, Public Health and Nursing, Universitas Gadjah Mada/Dr. Sardjito General Hospital, Yogyakarta, Indonesia.
*Corresponding Author : Galuh Martin Maytasari
Department of Child Health, Faculty of Medicine, Public
Health and Nursing, Universitas Gadjah Mada/Dr. Sardjito
General Hospital, Yogyakarta, Indonesia.
Tel: +62-813-932-27962; Email: galuhmartin@gmail.com
Received : Apr 04, 2022
Accepted : May 02, 2022
Published : May 09, 2022
Archived : www.jjgastro.com
Copyright : © Maytasari GM (2022).
Abstract
Background: Caroli’s disease is a rare congenital disease of the liver, characterized by cystic dilatation of the intrahepatic bile duct. There are two types of Caroli’s disease. First, classic Caroli’s disease involves malformation of the biliary tract alone, whereas Caroli’s syndrome refers to the presence of associated congenital hepatic fibrosis. The prevalence of both cases is less than 1: 1,000,000 inhabitants. The early diagnosis and treatment can be challenging as the disease is dynamic and progressive.
Case illustration: We present three cases of Caroli’s disease in tertiary level hospital. All patients had portal hypertension, which developed as a result of congenital hepatic fibrosis or bile duct dilatation. Two patients had congenital hepatic fibrosis and ascites, which one of them underwent peritoneal external drainage for managing refractory ascites. One patient had gastroesophageal varices that required endoscopic band ligation routinely. Multiple cyst kidney was reported in one patient. Another patient had Myelodysplastic Syndrome (MDS). Each patient had different imaging modality to establish the diagnosis. Liver biopsy conducted in one patient showed the appearance of liver fibrosis. They received supportive care for the portal hypertension and chronic cholestasis.
Summary: The diagnosis of Caroli’s disease is difficult without imaging study since no typical symptom, sign or laboratory examination were able to distinguish from other diseases. The treatment depends on the clinical features and the location of biliary abnormalities. Liver transplantation is considered in Caroli’s disease with liver fibrosis.
Keywords: Caroli’s disease; Caroli’s syndrome; Liver fibrosis; Children.
Citation: Maytasari GM, Widowati T. Caroli’s disease in children: A case series. Japanese J Gastroenterol Res. 2022; 2(7): 1080.
Introduction
Caroli’s disease is a rare congenital disease of the liver characterized by cystic dilation of the intrahepatic bile duct [1] First reported in 1958 by Jacques Caroli. There are two classification of this disease. Classic Caroli’s disease involves malformation of the biliary tract alone. This rare isolated form occurs only in about 15% cases. Caroli’s syndrome refers to the presence of associated periportal fibrosis (congenital hepatic fibrosis). This second form is more frequent [2]. Caroli’s disease is also classified by Todani et al [3] as type V choledochal cyst. Diffuse form affecting both lobes of liver is the most common form of Caroli’s disease. Monolobar involvement is more uncommon, and when present involves the left lobe more often than the right. Caroli’s disease usually presents in childhood and adolescent, but manifestation neonatal patients has also been reported [1].
Case 1
A 2-year-old female presented with jaundice and whiteclayed stool. She was referred from private hospital to our hospital for further investigation of cholestasis etiology. Upon first admission, the patient presented with jaundice, white-clayed stool, and developmental delayed. From physical examination, there were icteric sclerae, enlarged liver (liver span 12 cm) and spleen (Schuffner 2), microcephaly, and marasmic type severe malnutrition. Ascites was found as the disease progress. Cardiac and pulmonary examination showed no abnormality.
Laboratory examination showed anemia, cholestasis, hypoalbuminemia, prolonged of clotting time, elevated transaminase, elevated of gamma GT, dyslipidemia, vitamin D deficiency and positive CMV antigenemia. A doppler abdominal ultrasound was performed, which revealed intrahepatic multiple cysts, hepatomegaly with the appearance of liver fibrosis and signs of portal hypertension. From abdominal magnetic resonance imaging (MRI) with contrast showed choledochal cyst type V (according to Todani’s classification) (Figure 1) and biliary atresia (possibly type I), left kidney hypoplasia with multiple cysts, and hepatosplenomegaly.
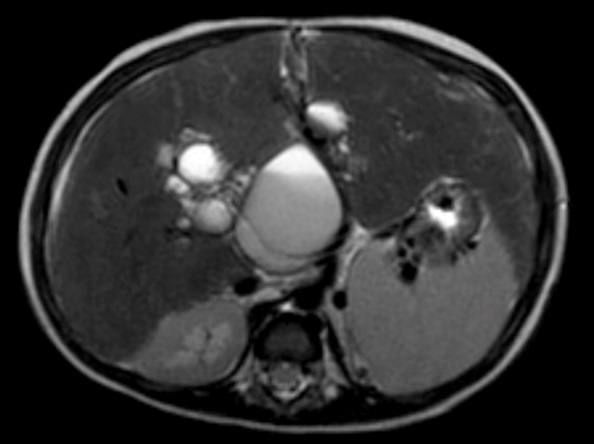
The patient has been admitted in our hospital for five times. Upon first admission, she was received ganciclovir as the treatment of CMV disease. Ursodeoxycholic acid (UDCA), propranolol, rifampicin, Vitamin A, D, E, K, and supportive treatment were given. The parents had been counseled and asked to come for regular follow up.
Case 2
A 6-month-old male came to a tertiary hospital with chief complaint of abdominal distention. He looked jaundice and had white clayed stool since one and a half month old. At 3 months old he was brought to a hospital. Liver biopsy was conducted in previous hospital, which revealed extrahepatic cholestasis and liver fibrotic without any sign of liver cirrhosis.
On physical examination, there were icteric sclerae, pallor conjunctival, caput medusae, abdominal distention and ascites. His liver edge was palpable 6.5 cm below right arch costae, and spleen was palpable at Schuffner II. There was also swelling in the scrotum. He was marasmic type severe malnutrition. Laboratory examination revealed normocytic normochromic anemia, cholestasis, normal ALT, and abnormality of liver function indicators such as hypoalbuminemia, prolonged clotting time (PPT and APTT). He was seronegative for HBsAg.
A vascular doppler sonography showed decreased portal venous velocity which indicate portal hypertension. Magnetic resonance cholangiopancreatography (MRCP) disclosed multifocal cystic dilatation of intrahepatic bile ducts, the common bile duct was undetectable appearance suggestive of liver cirrhosis, and splenomegaly (Figure 2).
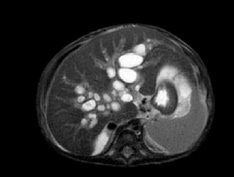
Peritoneal drainage was conducted due to massive ascites. UDCA, Vitamin A, D, E, K and spironolactone were administered.
Case 3
A 10-year-old male presented to a private hospital with recurrent blood vomiting since 4 years prior, and referred to our tertiary hospital for further evaluation. There were not any abdominal pain and yellowish skin reported between the complaint. Before the admission on the ward, he was referred to our hematology-oncology outpatient clinic for the complaint of pallor and need for repeated blood transfusion. He was suspected with thalassemia.
On physical examination he was found non-icteric, pallor conjunctival, his liver edge was 6 cm from the right costal margin in the mid clavicular line, and spleen was palpable at Schuffner II. Liver consistency was firm, and it was not tender. His nutritional status was moderate malnutrition and stunted. Other physical signs were unremarkable. Laboratory investigation showed microcytic hypochromic anemia, leukopenia, and thrombocytopenia. Peripheral blood smear revealed anemia with abnormal erythrocyte morphology, increased erythropoietic response, leukopenia with reactivity of neutrophil, and thrombocytopenia. Iron status indicators was low ferritin, low transferrin saturation, normal iron and TIBC. Bone marrow puncture was conducted, which showing increased erythropoiesis and thrombopoiesis response, dysplasia of erythropoiesis and granulopoiesis, myeloblast 5% and lymphoblast 4% was found. This result indicted possible of myelodysplastic syndrome. Liver function test remain stable with the following values: normal ALT and AST, normal albumin level, normal bilirubin, mild prolongation of APTT and PPT, and normal of serum albumin concentration.
The patient was seronegative for HBsAg.
A vascular doppler sonography disclosed multiple cysts in right liver lobe, splenomegaly and widened caliber of portal venous which indicate portal hypertension. An abdominal CT scan with contrast was also conducted. Hepatomegaly, dilatation of intrahepatic biliary duct particularly at the right lobe, the largest cyst measured 5,47 x 5,77 x 5,54 cm, suggested Choledochal cyst type V (Caroli’s disease, according to Todani’s classification), cholangitis and cholecystitis were found (Figure 3). There were also splenomegaly and right nephrolithiasis.
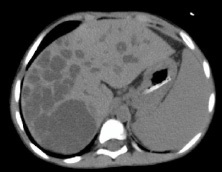
Upper gastrointestinal endoscopy (Figure 4) demonstrated the presence of grade 3-4 gastroesophageal varices, from corpus gastric proximally to the four columns of esophagus without active bleeding. Gastric mucosa was hyperemic, which indicate gastropathy. Ligation was done twice, two months apart.
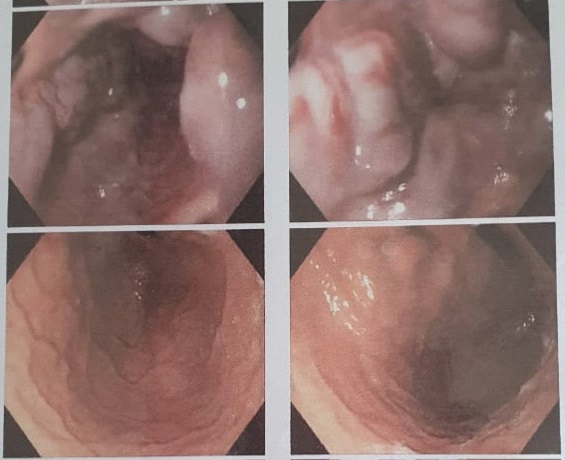
Table 1:
|
Case 1 |
Case 2 |
Case 3 |
Sex |
female |
male |
Male |
Age at diagnosis |
2 years old |
6 months old |
10 years old |
Clinical features |
Jaundice |
Jaundice |
Hematemesis |
Associated disease |
Polycystic kidney disease |
Bilateral hydrocele |
Myelodysplastic syndrome |
Nutritional status |
Marasmic type severe malnutrition |
Marasmic type severe malnutrition |
Stunted |
Laboratory examination |
Anemia |
Anemia |
Pancytopenia |
Imaging studies |
Doppler Abdominal ultrasound: intrahepatic multiple cysts, hepatomegaly, appearance of liver fibrosis, signs of portal hypertension, |
Doppler abdominal ultrasound: decreased portal venous velocity, 8 m/s |
Doppler abdominal ultrasound: multiple cysts in right liver lobe, splenomegaly and widened caliber of portal venous which indicate portal hypertension |
Treatment |
Ganciclovir for CMV infection |
Peritoneal external drainage |
Endoscopic band ligation |
Propranolol and omeprazole were given. He was planned to have another endoscopic variceal ligation in 1-2 months and regular follow up.
Discussion
Caroli described two forms of congenital dilatation of intrahepatic biliary tree associated with renal cystic disease. In the more common type, the portal lesion is the ductal plate malformation (DPM) type of congenital hepatic fibrosis (CHF). This entity is now referred to as Caroli’s syndrome. The second, much more rare type is characterized by pure ductal ectasia and is now called Caroli’s disease [4]. Both cases are extremely rare with an approximate prevalence of less than one in 1,000,000 inhabitants [5]. Caroli’s disease is also classified by Todani [3] as type V choledochal cyst. Diffuse form that affecting both lobes is the most common form. When presented monolobar, left lobe involvement is more common than right lobe. This condition often presents between the ages of 5 and 20 years, [1] though neonatal cases has also been reported before, and more common in female, with a male to female ratio of 1: 1.8 [4,5].
The embryonic basis for the development of Caroli’s disease is the ductal plate malformation leading to persistent embryonic bile ducts. DPM is caused by in utero vascular accident. DPM of the larger intrahepatic bile ducts lead to the development of Caroli’s disease, whereas congenital hepatic fibrosis is a result of malformations at the level of the interlobular bile ducts [1]. Autosomal recessive inheritance is most commonly proposed, but the autosomal-dominant inheritance has also been reported [1,4].
Caroli’s disease has been associated polycystic kidney disease, renal medullary spongiosis and medullary cystic disease [5]. Mutation in the PKD1 and PKD2 are thought to participate in this condition. Lawrence-Moon-Biedl syndrome and systemic amyloidosis have also been reported in patient with Caroli’s disease [4]. Presenting sign and symptoms are intermittent abdominal pain, hepatomegaly, jaundice and steatorrhea due to bile stagnation. In Caroli’s syndrome, due to congenital hepatic fibrosis, sign of portal hypertension is present, and esophageal varices may developed [4]. As the disease progress, cholangitis, cholelithiasis, biliary abscess, septicemia, and cholangiocarcinoma as potential complications can occur. Its clinical manifestation highly variable and may appear early or late during life [6].
The laboratory findings are non-specific. The complete blood count may reveal thrombocytopenia and leukopenia if portal hypertension and hypersplenism are present. Anemia presents in hematemesis and melena. Elevated white blood cell and erythrocyte sedimentation rate may indicate cholangitis. Transaminase levels may be slightly elevated. Elevated BUN and creatinine present in associated renal disease [6].
The gold standard for diagnosis of Caroli’s disease is Cholangiography. Magnetic resonance cholangiopancreatography (MRCP) is emerging as the modality of choice for diagnosis. Park et al [7] reported good accuracy of MRCP to detect and classify the choledochal cyst and revealed associated cholangiocarcinoma and choledocholithiasis. Other imaging studies are abdominal MRI, abdominal CT, ultrasonography, and isotope scans [1,8]. The treatment of Caroli’s disease depends on the clinical features and the location of the biliary abnormalities.
Ursodeoxycholic acid (UDCA) is used for treating and preventing future episode of hepatolithiasis and cholangitis. Antibiotic is needed for cholangitis as the most complication of Caroli’s disease. Beta-blockers are recommended in portal hypertension. Variceal bleeding in can be prevented with endoscopic band ligation or sclerotherapy. Surgical resection (partial hepatectomy, lobectomy) is an option for segmental or unilobar involvement. Liver transplantation is considered for diffuse form or multilobar involvement and in congenital hepatic fibrosis of Caroli’s syndrome. However, patients with associated congenital hepatic fibrosis had worse survival. No consensus has been reached on the indication or timing of liver transplantation with Caroli’s disease. Habib et al [9] reported the majority patients of liver transplantation were signs of hepatic decompensation like ascites, encephalopathy, coagulopathy, portal hypertension, jaundice, prolonged prothrombin time, or decreased albumin. Other series reported recurrent cholangitis as the primary indication for liver transplantation [10,11].
Conclusion
In summary, Caroli’s disease is a rare congenital malformation of the intrahepatic bile duct. The clinical manifestation is highly variable. Hence, imaging studies are needed to established the diagnosis, with MRCP being choice if cholangiography is unavailable. Medical therapy given depends on its clinical features and complications. Surgical intervention to liver transplantation is also considered as the treatment.
References
- Ananthakrishnan AN & Saeian K. Caroli’ s Disease : Identification and Treatment Strategy. 2007.
- Besser P, Bacia V, Mazur W & Gonciarz Z. Caroli’ s disease : case report and review of the literature. 2003; 4: 176-181.
- Todani T. Congenital bile duct cysts Classification, operative procedures,choledochal cyst Am J Surg. 1977; 134: 263-269.
- Suchy FJ, Sokol RJ & Balistreri WF. Liver Disease in Children. (Cambridge University Press) 2007.
- Correia PC & Morgado B. Caroli’ s Disease as a Cause of Chronic Epigastric Abdominal Pain : Two Case Reports and a Brief Review of the Literature. 2017; 9.
- Yonem O & Bayraktar Y. Clinical characteristics of Caroli’s syndrome. 2007; 13: 1934-1937.
- Park DH, et al. Can MRCP replace the diagnostic role of ERCP for patients with choledochal cysts? Gastrointest. Endosc. 2005; 62: 360-366.
- Tajik P & Goudarzian AH. Caroli Syndrome in a Child: A Case Report. Case Reports Clin. Pract. 2019; 4: 89-93.
- Habib S, et al. Caroli’s Disease and Orthotopic Liver Transplantation. Liver Transplant. 2007; 13: 767-768.
- Sans M, et al. Liver transplantation in patients with Caroli’s disease and recurrent cholangitis. Transpl. Int. 1997; 10: 241-244.
- De Kerckhove L, et al. The place of liver transplantation in Caroli’s disease and syndrome. Transpl. Int. 2006; 19: 381-388.